
Comparison of Gene Selection and Machine Learning for
Tumor Classification
Qingzhong Liu
1
, Andrew H. Sung
1, 2
, Bernardete M. Ribeiro
3
1
Department of Computer Science
2
Institute for Complex Additive Systems Analysis
New Mexico Tech, Socorro, NM 87801, USA
3
Department of Informatics Engineering
University of Coimbra
3030-290 Coimbra, Portugal
Abstract.
Class prediction and feature selection are two learning tasks that are
strictly paired in the search of molecular profiles from microarray data. In this
paper, we apply the recursive gene selection proposed in our previous paper to
six types of micaroarray gene expression data for tumor classification. In com-
parison with other two well-known gene selections, SVM-RFE (Support Vector
Machine Recursive Feature Elimination) and T-test, our method outperforms
best. The kernel type and kernel parameters are critical to the classification per-
formances for the kernel classifiers. Our experiments indicate that RBF kernel
classifiers are pretty good under low feature dimensions; their performances in-
crease initially and then decrease as the feature dimension increases.
1 Introduction
Microarrays are capable of profiling the gene expression patterns of tens of thousands
of genes in a single experiment. DNA targets are arrayed onto glass slides (or mem-
branes) and explored with fluorescent or radioactively labeled probes [1]. Obtaining
gene expression data from cancerous tissues gives insight into the gene expression
variation of various tumor types, thus providing clues for cancer classification of
individual samples. One of the key challenges of microarray studies is to derive bio-
logical insights from the unprecedented quantities of data on gene expression pat-
terns. Partitioning genes into closely related groups has become an element of practi-
cally all analyses of microarray data [2]. However, identification of genes is faced
with many challenges. The main challenge is the overwhelming number of genes
compared to the smaller number of available training samples. In machine learning
terminology, these data sets have high dimension and small sample size. Many of
these genes are irrelevant to the distinction of samples. These irrelevant genes have a
negative effect on the accuracies of the classifier. Another challenge is that DNA
array data contain technical and biological noises. Thus, it is critical to identify a
subset of informative genes from a large data pool that will give higher classification
Liu Q., H. Sung A. and M. Ribeiro B. (2006).
Comparison of Gene Selection and Machine Learning for Tumor Classification.
In Proceedings of the 2nd International Workshop on Biosignal Processing and Classification, pages 13-22
DOI: 10.5220/0001219700130022
Copyright
c
SciTePress

accuracy.
Because DNA microarray data has high dimension and small samples, the gene
selection is very important to the classification accuracy. T-TEST [3] is one well-
known gene selection in DNA microarray analysis. It ranks the significant genes
according to the p-values. Unfortunately, it just considers the individual gene, not the
interaction of genes. And the problem probably is that we might end up with many
highly correlated genes. If there is a limit on the number of genes to choose we might
not be able to include all informative genes. The method in [4] is to retrieve groups of
similar genes, and apply a test-statistic to select genes of interest. SVM-RFE (Support
Vector Machine Recursive Feature Elimination) is another well-known gene selection
which refines the optimum feature set by using SVM in a wrapper approach [5].
Peng, Long and Ding [6] presents a feature selection to achieve the max-dependency,
max-relevance, and min-redundancy based on mutual information. Recently, a
scheme of gene selection based on recursive feature addition and similarity measures
between the chosen genes and the candidates [7]. In comparison with the well-known
gene selections, T-TEST and SVM-RFE using different classifiers, on the average,
the method of recursive gene selection is the best regarding the classification accu-
racy under different feature dimensions.
In this paper, we apply the three gene selections of recursive feature addition [7],
SVM-RFE [5] and T-test. We compare the
mean test accuracy and the highest test
accuracy under the highest train accuracy, and the highest test accuracy in the ex-
periments.
Additionally, we apply several learning classifiers to the experiments, and
compare the classification performances on the gene selections. Generally, in the
three gene selection, recursive gene addition performs the best; in the learning classi-
fiers, on the average, Nearest Mean Scale Classifier (NMSC) and kernel classifiers
with polynomial kernels have better classification accuracy than others. Under the
high feature dimensions, the classification performances of RBF kernel classifiers
decrease along with the increase of the feature dimension.
2 Recursive Feature Additions for Gene Selection
Liu and Sung [7] proposed a scheme of gene selection based on supervised learning
and similarity measure between chosen genes and candidates. We described it in brief
as follows:
1. Insignificant or noise microarray gene data is filtered out according to test-
statistical selection.
2. Each individual gene is ranked in the order from the highest classification accu-
racy to the lowest classification accuracy with some classifier.
3. The gene with the highest classification accuracy is chosen as the most important
feature, or the first feature. If there are many genes with the same highest classifica-
tion, the most important feature is set to the gene with the lowest p-value measured by
test-statistic. At this point the chosen feature set, G
1
, consists of the first feature, g
1
,
which corresponds to feature dimension one.
4. The N + 1 – dimension feature set, G
N+1
= { g
1,
g
2
,…, g
N
, g
N+1
} is produced by
14

adding g
N+1
into the chosen N-dimension feature set, G
N
= { g
1,
g
2
,…, g
N
}. The
choosing of the N+1
th
feature g
N+1
is described as follows.
Each gene g
i
(i ≠ 1, 2, …, N) outside of G
N
is added into G
N
; the classification ac-
curacy of each feature set G
N
+ {g
i
} is compared, the g
c
with the highest classifica-
tion accuracy is marked and put into the set of candidates, C. Generally, the set of
candidates consists of many genes, but only one gene in the set of candidates will be
the chosen. Three strategies are designed for choosing the g
N+1
:
The first strategy is to compare the individual classification accuracy of the candi-
dates. The candidates with the highest accuracy will be put into the final stunt; the
gene with the lowest p-value in the final stunt is chosen as the N+1
th
feature g
N+1
.
The second strategy is to measure the similarity of chosen genes and candidate
genes. Pearson’s correlation [19] between the chosen gene g
n
(g
n
∈
G
N
, n = 1, 2…
N) and the candidate g
c
(g
c
∈
C, c= 1,2 … m; m is the number of the elements in C.)
is calculated. The sum of the square of the correlation (SC) is calculated to measure
the relation, defined as follows:
SC(g
c
) =
∑
cor
=
N
n 1
2
(g
c
, g
n
) (1)
Where, g
c
∈
C, g
n
∈
G
N
, n = 1, 2… N. The g
c
with the minimal value of SC(g
c
) is
chosen as g
N+1
.
The third strategy is to calculate the maximum value of the square of the correla-
tion (MC),
MC(g
c
) =
ma
x
(cor
2
(g
c
, g
n
) ), n = 1, 2… N. (2)
Where, g
c
∈
C, g
n
∈
G
N
. The g
c
with the minimum value of MC(g
c
) is chosen as
g
N+1
.
In the methods mentioned above, a feature is recursively added into the feature set
under the supervised learning. Similar to the name of SVM-RFE, we call the strate-
gies Classifier-Recursive Feature Addition (C-RFA), Classifier Minimal Sum of the
square of Correlation-Recursive Feature Addition (CMSC-RFA), Classifier Minimal
Maximal value of the square of Correlation-Recursive Feature Addition (CMMC-
RFA), respectively. For example, if the classifier is Naïve Bayes Classifier (NBC),
we record them NBC-RFA, NBCMSC-RFA, and NBCMMC-RFA, respectively.
3 Experiments
3.1 Data Sets
The following benchmark datasets are tested in our experiments. If the data source is
not mentioned, it is available at:
http://www.broad.mit.edu/cgi-bin/cancer/datasets.cgi.
1) The LEUKEMIA data set consists of two types of acute leukemia: 48 acute lym-
phoblastic leukemia (ALL) samples and 25 acute myeloblastic leukemia (AML)
samples, over 7129 probes from 6817 human genes. It was originally studied in the
15

paper [8].
2) The LYMPHOMA data set consists of 58 diffuse large B-cell lymphoma (DLBCL)
samples and 19 follicular lymphoma (FL) samples. It was studied in the paper [9].
The data file, lymphoma_8_lbc_fscc2_rn.res, and the class label file, lym-
phoma_8_lbc_fscc2.cls are used in our experiments for identifying DLBCL and
FL.
3) The PROSTATE data set in the paper [10] contains 52 prostate tumor samples and
50 non-tumor prostate samples.
4) The COLON data set in the paper [11] contains 62 samples collected from colon-
cancer patients. Among them, 40 tumor biopsies are from tumors and 22 normal
biopsies are from healthy parts of the colons of the same patients. 2000 genes were
selected based on the confidence in the measured expression levels. The data
source is available at:
http://microarray.princeton.edu/oncology/affydata/index.html.
5) Only dataset C in the dataset of Central Nervous System (CNS) embryonal tumor
[12] that is used to analyze the outcome of the treatment contains 60 patient sam-
ples, 21 are survivors who are alive after treatment and 39 are failures who suc-
cumbed to their diseases. There are 7129 genes.
6) Breast cancer dataset associated with the paper [13] contains 97 patient samples,
46 patients are relapse who had developed distance metastases within 5 years, and
51 patients are non-relapse who remained healthy from the distance after their ini-
tial diagnosis for interval of at least 5 years. The data source is available at:
http://www.rii.com/publications/2002/vantveer.htm.
3.2 Experimental Setup
Our experiments are described as follows:
1. The training samples are chosen from the datasets at random. The rest samples are
used for test. The ratio of training sample to test sample is 3:2 in the same class.
2. The gene selections, T-TEST, SVM-RFE, and the Recursive Feature Additions are
applied for gene selection based on the training samples. Different feature sets of
the gene expression data are produced under the feature dimension 1 to 50.
3. Several classifiers are applied to the feature sets extracted from test samples.
4. Repeat steps 1-3 30 times.
Although the results in [7] show that, regarding the average test accuracy in each
feature dimension, overall, the recursive gene addition performs best, the statistic for
evaluation is not enough. In our new experiments, the following statistics are meas-
ured to evaluate the performances of the gene selections.
(1) The average test accuracy under the condition that the associated train accuracy is
the highest in the experiments.
(2) The highest test accuracy under the condition that the associated train accuracy is
the highest in the experiments.
(3) The highest test accuracy in the experiments.
16
In our experiments, Naive Bayes Classifier (NBC) is applied for recursive gene
addition. The test classifiers are Nearest Mean Scale Classifier (NMSC), Kernel
Fisher Discriminant (KFD), Support Vector Machine (SVM), NBC, and uncorrelated
normal based quadratic Bayes Classifier that is recorded as UDC [14, 15, 16, 17, 18].
3.3 Comparison of Gene Selections
3.3.1 Comparison of the Average Test Accuracy and the Highest Test Accuracy
Under the Condition that the Associated Train Accuracy is the Highest
Generally, the feature set that is associated with the highest train accuracy is prone to
be treated as the final feature set. We compare the mean test accuracy and the highest
test accuracy under the condition that the associated train accuracy is the highest.
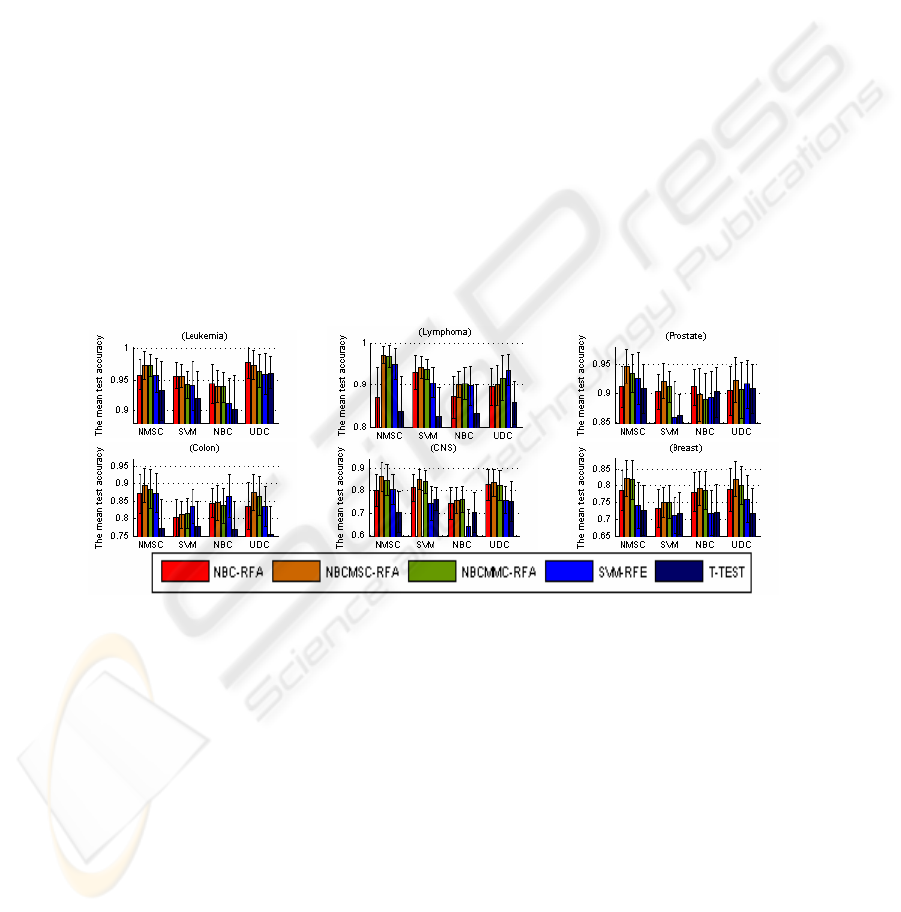
Fig. 1 lists the average test accuracy associated with the highest train accuracy. Fig.
1 indicates that, NBCMSC-RFA is the best, followed by NBCMMC-RFA, SVM-RFE
and NBC-RFA. On the average, T-TEST is not better than others. Fig. 2 lists the
highest test accuracy associated with the highest train accuracy in the experiments.
Fig. 2 also indicates that NBCMSC-RFA is the best, followed by NBCMMC-RFA,
SVM-RFE, and NBC-RFA. T-TEST is the least one. Because of the space limit, the
classification with the use of KFD is not shown in the figures.
Fig. 1. The mean test accuracy associated with the highest train accuracy. In the 24 groups,
NBCMSC-RFA is the best in 16 groups; NBCMMC-RFA is the best in the 4 groups (applying
NMSC to Leukemia, NBC to Lymphoma and CNS, and SVM to Breast); SVM-RFE is the best
in the 3 groups (applying UDC to Lymphoma, SVM to Colon, and NBC to Colon); and NBC-
RFA is the best in 3 groups (applying NBC and UDC to Leukemia, NBC to prostate). The
highest test accuracy is obtained by recursive gene selection based on the correlation measures
in the five types of cancer data (Lymphoma, Prostate, Colon, CNS, and Breast) and NBC-RFA
gains the highest test accuracy for Leukemia data classification.
17
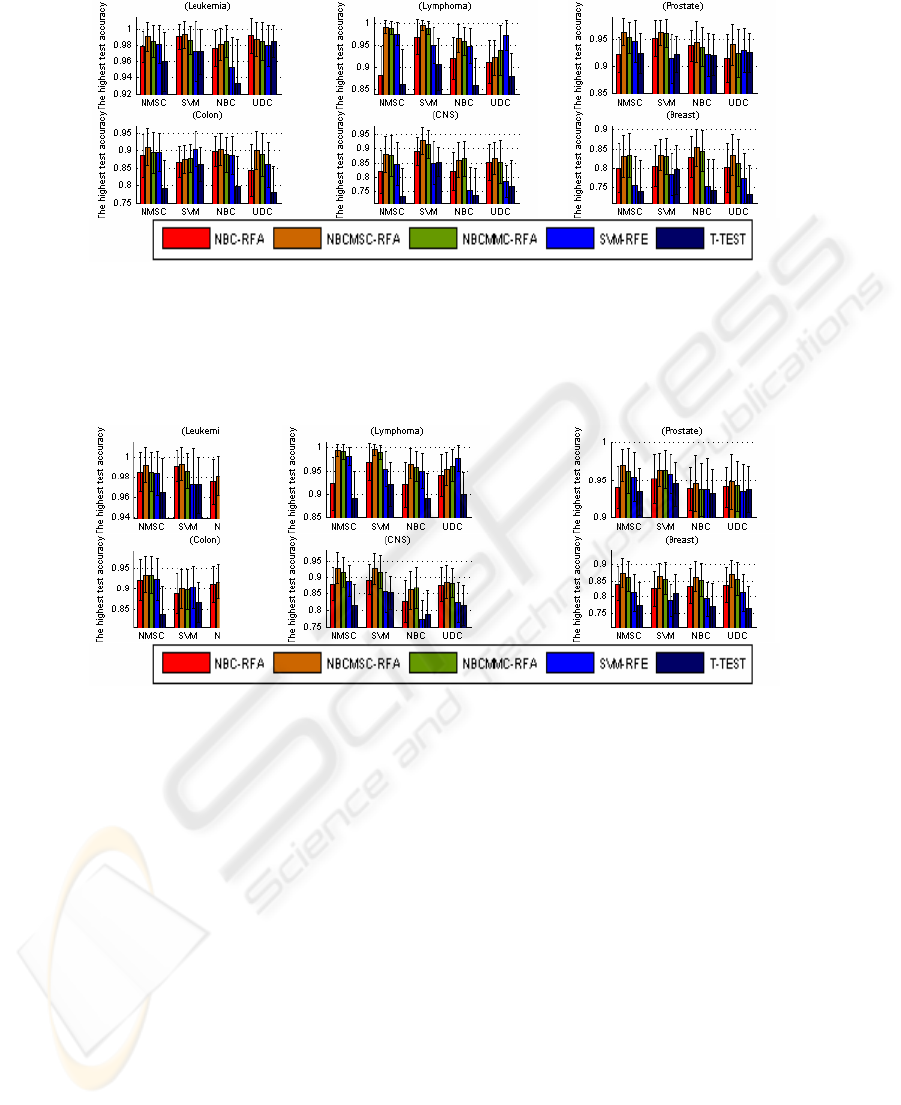
Fig. 2. The highest test accuracy associated with the highest train accuracy. In the 24 groups,
NBCMSC-RFA is the best in the 18 groups; NBCMMC-RFA is the best in the 3 groups (apply-
ing NBC to Leukemia and CNS, NMSC to Breast); SVM-RFE outperforms others in 2 groups
(applying UDC to Lymphoma, and SVM to Colon). NBC-RFA and T-TEST gain no champi-
onship. For the six types of cancer data, all the highest test accuracy is obtained by recursive
gene selection based on the correlation measures.
Fig. 3. The highest test accuracy under the feature dimension 1 to 5. In the 24 groups,
NBCMSC-RFA is the best in 19 groups; NBCMMC-RFA is the best in 3 groups; SVM-RFE is
the best in 2 groups (applying UDC to Lymphoma, SVM to Colon); NBC-RFA is the best in 1
group; and T-TEST gains no championship. In the test for each type of cancer data, all the
highest test accuracy is obtained by recursive gene selection based on the correlation measures
.
3.3.2 Comparison of the Highest Test Accuracy
Fig. 3 lists the highest test accuracy, regardless of the train accuracy. In the 24
groups, NBCMSC-RFA is the best in 19 groups; NBCMMC-RFA is the best in 3
groups; SVM-RFE is the best in 2 groups (applying UDC to Lymphoma, SVM to
Colon); NBC-RFA is the best in 1 group; and T-TEST gains no championship. In the
test for each type of cancer data, all the highest test accuracy is obtained by recursive
gene selection based on the correlation measures. It also indicates that NBCMSC-
RFA is the best, followed by NBCMMC-RFA, SVM-RFE, and NBC-RFA. T-TEST
is the worst in comparison with other gene selections. On the average, regarding
different classifiers, Figures 1 to 3 indicate that NMSC and SVM outperform others.
18
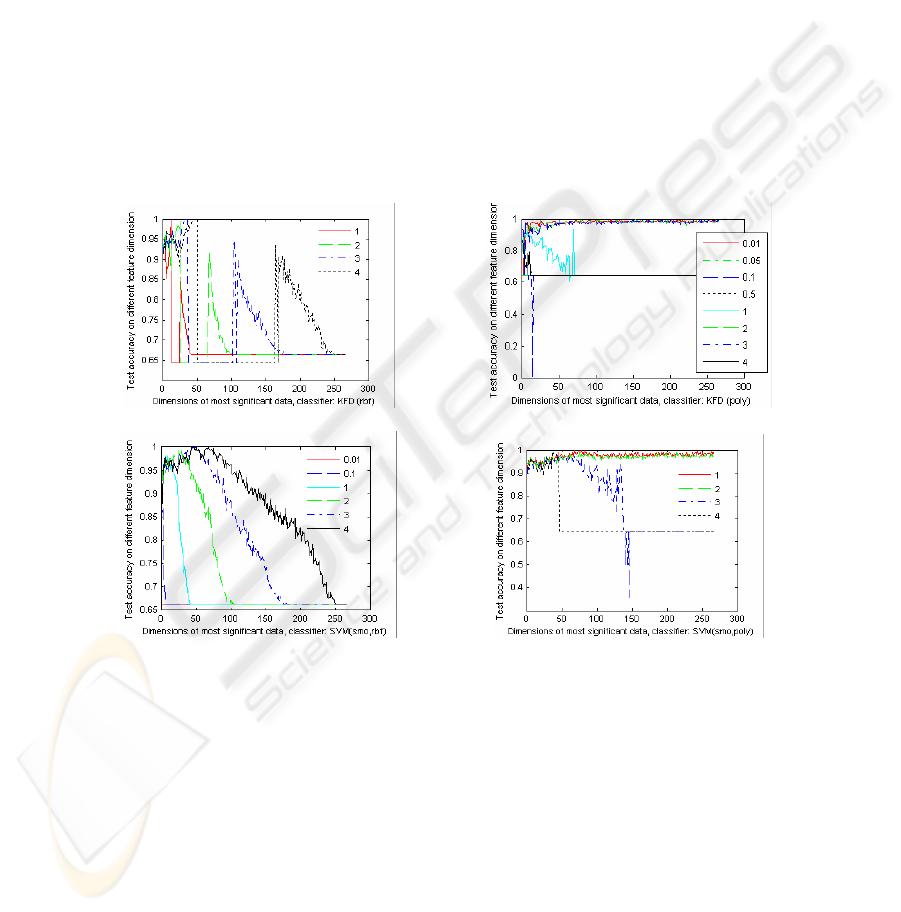
3.4 Comparison of Kernel Classifiers
Fig.4 - Fig. 6 compares the performances of the kernel classifiers, KFD and SVM
with different RBF kernel and POLY kernel for the gene selection of T-TEST under
different feature dimensions. Where, SMO (Sequential Minimal Optimization for
binary SVM with L1-soft margin) is applied to train model. The legends marked in
the Figures are the kernel arguments. The regularization of SVM is 10, and regulari-
zation of KFD criterion is 0.0001. Fig.4 - Fig. 6 indicates that the kernel type and the
kernel parameters are very important to the classification accuracy. They show that
the performances of the kernel classifiers, KFD and SVM with a RBF kernel will
increase initially as the feature dimension increases, and then decrease when the fea-
ture dimension increases continually. While the recognition performances of the
POLY kernel classifiers improve as feature dimension increases. The exception
shown in Fig. 4 (a) are resulted from the bad train models.
(a)
(b)
(c)
(d)
Fig. 4. The test classification of KFD with different RBF kernels (a) and POLY kernels (b) and
SVM with different RBF (c) and POLY (d) kernels for LEUKEMIA.
19

(a)
(b)
(c)
(d)
Fig. 5. The test classification of KFD with different RBF kernels (a) and POLY kernels (b) and
SVM with different RBF (c) and POLY (d) kernels for LYMPHOMA.
(a)
(b)
(c)
(d)
Fig. 6. The test classification of KFD with different RBF kernels (a) and POLY kernels (b) and
SVM with different RBF (c) and POLY (d) kernels for PROSTATE.
20

(a)
(b)
Fig. 7. The train accuracy of SVM with RBF kernels (a) and KFD with RBF kernels (b) for
PROSTATE.
Fig. 7 shows the train classification accuracy of the KFD and SVM with RBF ker-
nels. It show that the train accuracy values of the RBF kernels are pretty high, almost
100%. We infer that, from the difference between the train accuracy (Fig. 7) and the
test accuracy (Fig. 6 (a), Fig. 6(c)), in a high feature dimension, even the training
error (or empirical risk) is very low, does not imply a small expected value of the test
error (called risk) [14, 20].
4 Conclusions
On the average, the recursive feature addition based on supervised learning and corre-
lation measures is better than the well-known gene selection methods of SVM-RFE
and T-TEST regarding the
mean test accuracy and the highest test accuracy under the
highest train accuracy, and the highest test accuracy in the experiments.
Regarding the classifiers in our experiments, on the average, NMSC and SVM out-
perform others in the majority tests. And the kernel type and kernel parameters are
important to the classification performances of the kernel classifiers. The RBF kernel
classifiers are pretty good under low feature dimensions; their performances increase
initially and then decrease as the feature dimension increases. The classification per-
formances of the POLY kernel classifiers improve as the feature dimension increase.
Acknowledgements
Partial support for this research received from ICASA (Institute for Complex Addi-
tive Systems Analysis, a division of New Mexico Tech) is gratefully acknowledged.
21

References
1. Brown,P. and Botstein,D. (1999) Exploring the New World of the Genome with DNA
Microarrays. Nature Genetics Supplement, 21, 33-37.
2. Quackenbush,J. (2001) Computational Analysis of Microarray Data, Nature Rev. Genetic,
2, 418-427.
3. Armitage,P. and Berry,G. (1994) Statistical Methods in Medical Research, Blackwell.
4. Jaeger,J., Sengupta,R. and Ruzzo,W.(2003) Improved Gene Selection for Classification of
Microarray, Pacific Symposium on Biocomputing 8, 53-64.
5. Guyon,I, Weston, J., Barnhill,S. and Vapnik,V. (2002) Gene Selection for Cancer Classifi-
cation using Support Vector Machines, Machine Learning, 46(1-3), 389-422.
6. Peng,H, Long,F. and Ding, C. (2005) Feature selection based on mutual information: crite-
ria of max-dependency, max-relevance, and min-redundancy, IEEE Trans. on Pattern
Analysis and Machine Intelligence, 27(8),1226-1238.
7. Liu,Q. and Sung,A.H. (2006) Recursive Feature Addition for Gene Selection, Proc. of
IEEE - IJCNN 2006, Vancouver, Canada.
8. Golub, T. et al. (1999) Molecular Classification of Cancer: Class Discovery and Class
Prediction by Gene Expression, Science, 286, 531-537.
9. Shipp,M. et al. (2002) Diffuse Large B-Cell Lymphoma Outcome Prediction by Gene
Expression Profiling and Supervised Machine Learning, Nature Medicine, 8(1), 68-74.
10. Singh,D. et al. (2002) Gene Expression Correlates of Clinical Prostate Cancer Behavior,
Cancer Cell, 1(2), 227-235.
11. Alon,U. et al. (1999) Broad Patterns of Gene Expression Revealed by Clustering Analysis
of Tumor and Normal Colon Tissues Probed by Oligonucleotide Arrays, Proc. Natl.
Acad.. Sci. USA, Cell Biology, 96, 6745-6750.
12. Pomeroy,S.L. et al. (2002) Prediction of Central Nervous System Embryonal Tumor
Outcome based on Gene Expression, Letters to Nature, Nature, 415, 436-442.
13. Van,L.J. et al.(2002) Gene expression profiling predicts clinical outcome of breast cancer,
Letters to Nature, Nature, 415, 530-536.
14. Vapnik,V. (1998) Statistical Learning Theory, John Wiley.
15. Schlesinger,M. and Hlavac,V. (2002) Ten Lectures on Statistical and Structural Pattern
Recognition, Kluwer Academic Publishers.
16. Heijden,F., Duin,R., Ridder,D. and Tax,D. (2004) Classification, Parameter Estimation
and State Estimation, John Wiley.
17. Taylor,J. and Cristianini,N. (2004) Kernel Methods for Pattern Analysis, Cambridge
University Press.
18. Webb,A. (2002) Statistical Pattern Recognition, John Wiley & Sons, New York.
19. Tan,P., Steinbach,M. and Kumar,V. (2005) Introduction to Data Mining, Addison-
Wesley, 76-79.
20. Scholkopf,B., Guyon,I., Weston,J. (2003) Statistical Learning and Kernel Methods in
Bioinformatics, Artificial Intelligence and Heuristic Methods in Bioinformatics, P. Fras-
coin and R. Shamir (Eds.) IOS press, 2003
22
