
Research on Obesity Caused by Genetic Defects in Specific
Populations
Zhiru Li
University of Washington Seattle WA, Seattle, U.S.A.
Keywords: Obesity Leptin, MC4R, UCP, β3 Adrenergic Receptors, PPARγ.
Abstract:
Obesity is caused by several factors, including diet, exercise, and the environment. However, one critical
factor that many people ignore is genetic abnormality. People are aware of the fact that the amount of food
ingested, and the amount of energy exerted can impact the formation of fat. Thus, obesity can be induced by
mutations or diseases in genes involved in these three systems in our body. And it is necessary to study the
problem of obesity caused by genetic defects. This paper focuses on 5 genetic problems to find the obesity
problems and solutions caused by genetic variants. It is found that Leptin and MC4R genes can increase
appetite and lead to obesity. And UCP and beta-adrenergic receptor during energy expenditure may result in
reduced energy expenditure or hormonal instability in the body, both of which will contribute to obesity. The
PPARy gene is essential for fat storage, and a mutation in the gene can result in an inability to store fat, which
will eventually lead to significant bodily damage. By studying and compiling the literature, this research has
identified the problems that arise after each genetic variation and the corresponding solutions.
1 INTRODUCTION
Genetic defects and obesity have received much
attention in the study of genes and metabolism in
different populations. Therefore, this paper
investigates the causes and solutions of obesity in
people with different congenital genetic defects.
Many genes have been discussed separately in the
context of congenital genetic defects which cause
obesity. But there was no definite study. This paper
focuses on the category and the study of the obesity
problem caused by genetic defects and divides the
basic elements of fat production in humans into three
major categories. The three categories are food
intake, energy expenditure, and fat storage. In each
section, one or two genetic defects were found to
demonstrate that congenital defects in genes cause
obesity.
Meanwhile, this study summarizes and lists the
possible obesity problems and solutions separately,
demonstrating the root cause for understanding the
common genetic defects that cause obesity problems.
Therefore, this paper systematically summarizes the
essential congenital genes that cause obesity through
clinical cases and mouse experiments and provides
solutions to the genetic defects as well as future
therapeutic prospects. Also, this study will make it
easier and more straightforward for researchers in the
field of genetic variants and congenital genetic
defects to understand the problems.
2 FACTORS THAT INFLUENCE
OBESITY
Table 1 lists the five genes that are crucial in obesity,
as well as the function of each gene. The function
of genes and genetic defect problems will be explored
in the following section.
212
Li, Z.
Research on Obesity Caused by Genetic Defects in Specific Populations.
DOI: 10.5220/0011196200003443
In Proceedings of the 4th International Conference on Biomedical Engineering and Bioinformatics (ICBEB 2022), pages 212-219
ISBN: 978-989-758-595-1
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
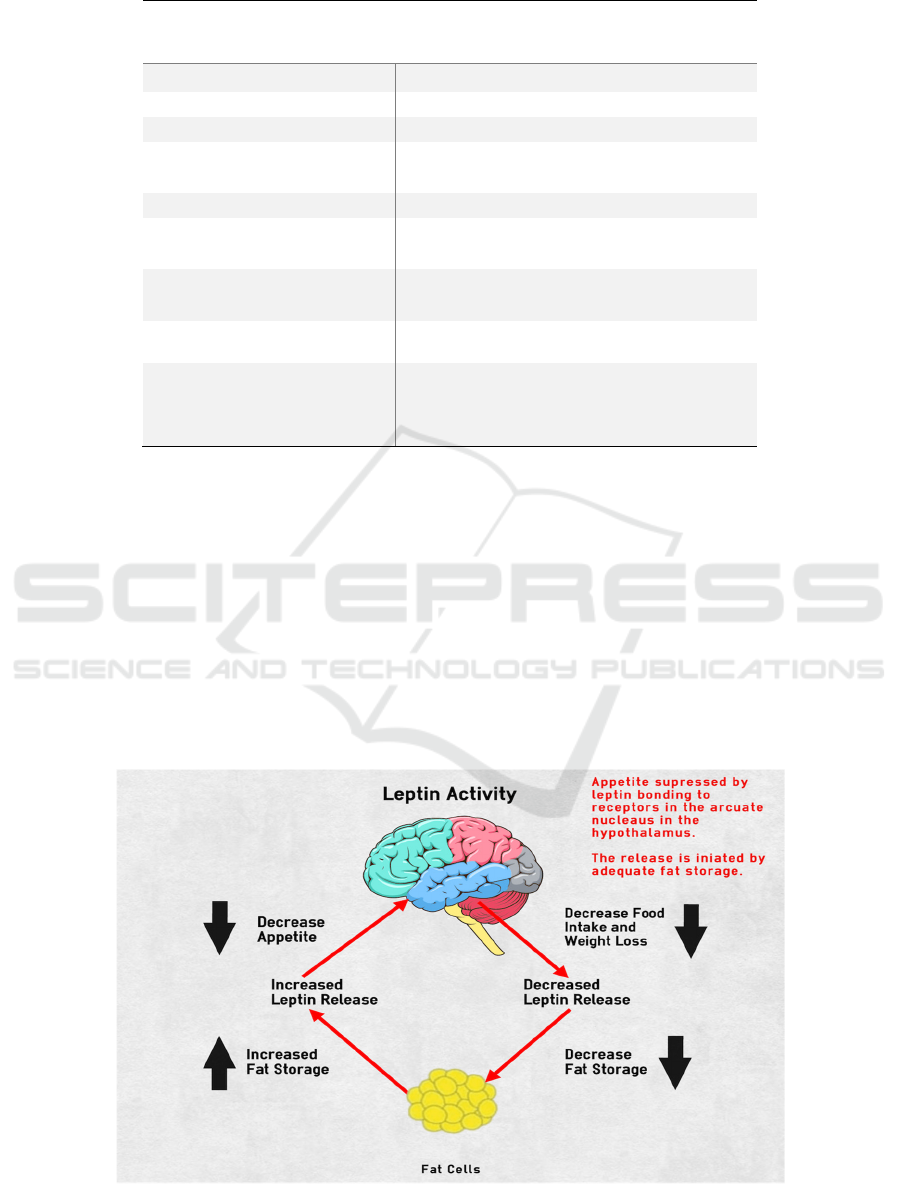
Table 1: Gene with CLEARLY defined role in obesity (Gao 2014)
GENE WITH CLEARLY
DEFINED ROLE IN
OBESITY
GENE NAME Function
FOOD INTAKE
LEPTIN Regulation eating behavior
MELANOCORTIN 4
RECEPTOR
Receptors of neuropeptides
ENERGY EXPENDITURE
UNCOUPING PROTEIN 1 Proton transporter and thermogenesis
ΒETA 3 ADRENERGIC
RECEPTOR
Lipolysis and thermogenesis in adipose tissue
FAT SYNTHESIS AN
STORAGE
PEROXISOME
PROLIFERATOR-
ACTIVATED RECEPTOR-
GAMA
Master gene of lipogenesis and adipogenesis
2.1
Genetic Defect Problems
2.1.1 Food Intake Leptin
Food intake usually depends on our appetite and
satiety; many genes control appetite and satiety, the
first and foremost being the leptin gene. Adipocytes
generate the hormone leptin, and leptin is the crucial
factor for regulates food intake and energy balance.
(Chen 2005) The leptin receptor is a protein that
leptin binds to and activates. The leptin receptor
protein is located on the cell surface of numerous
organs and tissues in the body, including the
hypothalamus which is a component of the brain. The
primary role of the hypothalamus is to control hunger
and thirst and also to control the quality of sleep and
mood. In the hypothalamus, leptin is a very important
hormone. Leptin binds to its receptors, triggering a
cascade of chemical signals that alter appetite and aid
in the production of satiety. Leptin, produced by fat
cells, regulates biological behavior and metabolism
by binding to central nervous system receptors.
Leptin deficiency or resistance can lead to severe
obesity in humans.
Figure 1: Leptin Activity (Chuck 2021).
Research on Obesity Caused by Genetic Defects in Specific Populations
213

This figure shows where leptin comes from and
how it works in the most basic way. Leptin is released
from adipose tissue, and the larger the number of
adipose tissues, the greater the amount of leptin
released, and vice versa. At the hypothalamic location
in the brain, leptin interacts with leptin receptors to
signal satiety. In other words, leptin acts in the
hypothalamus to control appetite based on fat storage
levels. According to the figure, as fat storage
increases, fat cells produce more leptin, which
suppresses hunger in the hypothalamus and causes
people to eat less. When people lose weight, their
leptin levels would drop, and their appetite would
returns. This graph depicts leptin activity in the
average individual. When a person has a congenital
leptin gene deficiency, fat cells do not release leptin
hormone, and leptin receptors in the hypothalamus do
not receive leptin hormone regardless of fatness, and
hence do not control appetite. Finally will lead to
severe obesity.
2.1.2 MC4R
In addition to the congenital deficiency of the leptin
gene, another gene that affects food intake leading to
obesity is the melanocortin-4 receptor gene (MC4R),
it is also considered to be the most common
monogenic form of obesity in humans (Chung 2012)
“MC4R is a single exon gene on chromosome
18q21.3. It encodes a G-protein coupled receptor
expressed mainly in the brain.” (Abdullah 2016) In
the hypothalamus, the melanocortin-4 receptor gene
(MC4R) is a key regulator of energy balance, food
intake, and body weight. (Doulla 2021) As mentioned
earlier, leptin acts mainly on neurons in the arcuate
nucleus of the hypothalamus and in the fasting state,
a decrease in leptin increases food intake by
stimulating agouti-related peptide (AgRP) neurons.
At the same time, leptin inhibits POMC neurons and
reduces the amount of alpha-MSH (melanocyte-
stimulating hormone), which normally inhibits food
intake. Second-order neurons with melanocortin-4
receptors (MC4R) synthesize and integrate these
signals.
The main clinical features of MC4R deficiency
are hyperphagia, increased appetite, and impaired
satiety. Adults with MC4R deficiency had
significantly lower blood pressure and a decreased
risk of hypertension than those of same age and
weight. (Melanocortin 2021) The heart rate and
sympathetic tone values are generally lower in
patients with MC4R deficiency. Patients with MC4R
deficiency have difficulty achieving weight loss
because people tend to be less responsive to diet and
exercise.
Complete deletion of MC4R also occurs and it is
similar to the symptoms of MC4R deficiency.
Abdullah, S and Reginold, W etc. reported a 4.5-year-
old child with a complete deletion of one copy of
MC4R. The young child presented obesity, tall
stature, overall developmental delays, and sexual
equality. (Abdullah 2016) The report also states that
the child weighed 7 pounds 4 ounces at birth and had
poor growth until 15 months of age when he began to
gain weight rapidly. He had gained a lot of weight by
the time he was 18 months old owing to his regular
overeating.
2.2 Energy Expenditure
2.2.1 UCP1
Human energy consumption depends on many factors
such as gender, age, and weight. A huge part of the
factors related to energy expenditure that leads to
obesity is due to the brown adipose tissue (BAT)
(Pravednikova 2020) and the uncoupling protein
(UCP) that are mainly found in the mitochondria of
brown cell tissues. Therefore, the study of the UCP
family can be of great help to the causes of obesity.
Uncoupling protein 1(CUP1) is a mitochondrial
membrane protein involved in brown adipocyte
adaptive thermogenesis. UCP1 is the only component
that is capable of transporting protons across the inner
membrane of mitochondria in brown adipocytes. In
this mechanism, UCP1 acts as a proton carrier that is
activated by free fatty acids, producing a shunt
between respiratory chain complexes and ATP
synthase. When UCP1 is active, the uncoupling
process results in a futile cycle and the loss of
oxidative energy as heat.
Theoretically, UCP1 deficiency should lead to a
decrease in fat burning capacity and then lead to
obesity. Kozak, L’s articles8 have proposed
experiments with mice because brown adipose tissue
is the primary source of heat supply in mice, and there
is little brown adipose tissue in adults that
corresponds to that of mice. Interestingly, it has been
shown from mouse experiments that UCP1
deficiency causes mice to be forced to use other
thermogenic, metabolic systems, so UCP1 deficiency
does not cause obesity in mice. In other words, UCP1
deficiency is not the leading cause of congenital
obesity in children or people. (Kozak 2005) However,
mice that lack UCP1 were more likely to develop fat
accumulation with age than normal mice in the
experiment. This experiment also proves that people
with UCP1 deficiency are more likely to develop
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
214

obesity as they age than the general population.
(Kontani 2005)
2.2.2 Beta 3-Adrenergic Receptors
β3 adrenergic receptors (β3-AR) are found on the cell
surface of both white and brown adipocytes and are
involved in lipolysis and thermogenesis. In humans,
β3-ARs are widely expressed in brown adipose
tissue, while white adipose tissue has few or no
receptors. The relationship between β3-ARs and
UCP1 is inextricably linked in terms of heat
production. Uncoupling protein -1 is primarily
responsible for the thermogenic activity of brown
adipose tissue. Hypothermia stimulates Ucp1 gene
expression via the sympathetic nervous system and -
adrenergic receptors. (Razzoli 2018)
Variants or absence of β3-ARs can lead to obesity
or less energy expenditure. (Clément 1970) Most of
the β3-ARs mutations are mutations in the Trp64Arg
of the β3-ARs, which can lead to an increased
probability of weight gain, insulin resistance, or
dyslipidemia, as well as a lower-body metabolic rate.
Defects in 3-adrenergic receptor binding, signaling,
or regulatory processes in obese people with such
mutations may result in reduced lipolytic reactions in
adipose tissue, aggravating obesity.
2.3 Fat Synthesis
The previous two parts examined the effect of genes
on appetite and thermogenesis on obesity, starting
from food intake and energy expenditure
respectively. The main direction of this paragraph is
about the relationship between fat storage and
production and obesity. (Lazar 2004)
In adipogenesis, peroxisome proliferator-
activated receptors γ are the primary regulators of
adipogenesis. The most abundant expression of
PPAR is found in adipose tissue, and two different
PPARγ isoforms, PPARγ1 and PPARγ2, have been
identified. Cells are also found in other organs, such
as breast, colon, liver, and vascular cells. The
expression of the PPARγ2 isoform appears to be
entirely adipocyte specific.(Stienstra 2007)
Adipogenesis is dependent on PPAR, and PPAR
target genes regulate adipocyte differentiation, lipid
storage, and glucose metabolism. In the liver, PPARγ
is able to balance triglycerides and helps contribute to
steatosis. At the same time, PPARγ in the liver
protects other tissues from triglyceride accumulation
and insulin resistance. In addition to this, PPARγ is
also involved in managing inflammatory responses
and has been shown to reverse macrophage
infiltration and thus reduce inflammatory gene
expression.
Therefore, PPARγ plays an essential role in our
bodies. When there is a deficiency or variation of
such an important gene, the body can suffer serious
consequences. This issue was confirmed in mouse
experiments, where mice's previous adipose-specific
knockdown of the PPARγ gene did not show a
dramatic phenotype in vivo. However, a specific
mouse type, Adipoq-Cre mice, was used to drive
adipose-specific recombination during the
experiments. At 3 months of age, there is little visible
brown and white adipose tissue, according to reports.
As a result, mice appeared to have hugely enlarged
pancreatic islets, massive fatty liver, and dramatically
increased blood glucose and serum insulin levels with
extreme insulin resistance. (Wang 2003) All of which
are major complications of obesity and common
pathophysiological bases for the development of
metabolic disorders and other chronic diseases in
obese populations.
3 TREATMENTS
3.1 Congenital Leptin Deficiency
(Leptin Injection)
Congenital leptin deficiency is a specific condition
that causes obesity and is relatively simple to treat.
Daily subcutaneous injections of recombinant human
leptin have been shown to be effective in treating
congenital leptin deficiency, resulting in persistent
beneficial effects on weight reduction, reduced
hunger, normal pubertal development, and
hyperinsulinemia, according to theoretical and
clinical instances.
During the treatment process, some
considerations need to be taken into account. Before
the injection may be administered, the patient must
first be clinically examined for an endocrine,
sympathetic, and immunological function to confirm
that they are normal. There are examples of clinical
use of recombinant methionyl human leptin (r-metHu
Leptin, Amylin Pharmaceuticals, San Diego, USA).
In addition to this leptin, injections need to be
administered at low physiological doses and need to
mimic the normal circadian rhythm of leptin, usually
starting at 6 pm. (Paz-Filho 2009) This article
confirmed that the body composition, food intake and
energy expenditure, lipid and glucose metabolism,
sympathetic tone, and gonadal, adrenal, somatic, and
thyroid functions are all affected by treatment.
Research on Obesity Caused by Genetic Defects in Specific Populations
215
Somatic and thyroid functions. The article also
mentions other conditions affected by leptin include
obesity, steatosis syndrome, diabetes, hypothalamic
amenorrhea, and anorexia nervosa. (Paz-Filho 2010)
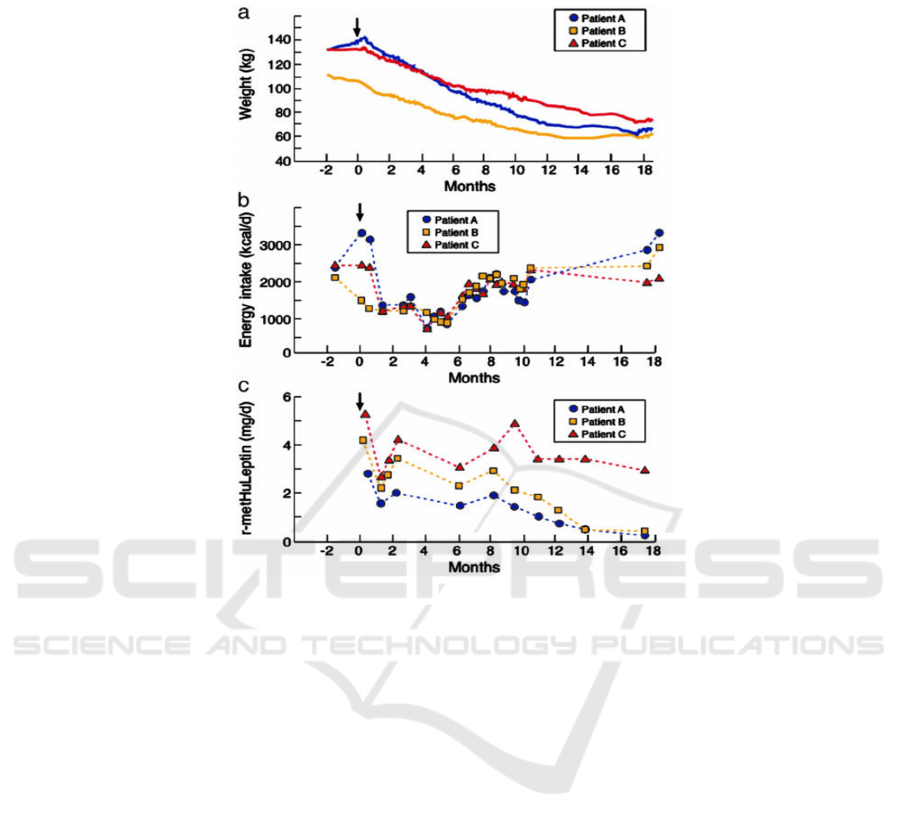
Figure 2: The effect of leptin treatment in adults (Licinio 2021)
These three graphs were produced by Julio
Licinio, Sinan Caglayan et al. for three leptin-
deficient adults treated with r-metHuLeptin injections
for 18 months. In Figure A, it can be seen that all three
patients had significant weight loss. During the
treatment process, the three patients lost weight
without any dietary changes or additional daily
activities. Figure B shows the energy intake of the
three patients. It shows a relationship between weight
loss and energy intake. R-metHuLeptin injections
suppressed their appetite and allowed them to lose
weight by reducing their energy intake. All three
individuals had significant weight loss after 18
months of treatment. Patient A, for instance, lost
more than half of his initial body weight. These
graphs reveal that leptin injections can effectively
cure patients with congenital leptin deficiency.
3.2
MC4R Deficiency (Bariatric
Surgery or Injections of
Liraglutide)
MC4R gene mutations, also called MC4R deficiency,
are divided into homozygous and heterozygous
mutations. There are corresponding treatments for
both mutations; Obesity is linked to heterozygous
MC4R mutations in a dominantly inherited manner.
MC4R deficiency affects around 2-5 percent of obese
children, 1% of obese adults, and about 1/500 of the
general population, making it the most prevalent
single-gene cause of obesity. (Melanocortin 2021)
The treatment for MC4R heterozygous mutations is
bariatric surgery, called Roux-en-Y-bypass surgery.
The size of the upper stomach will be reduced to a
tiny pouch roughly the size of an egg with this
operation. This will result in weight loss, however, in
those who have homozygous mutations, this
procedure may not be beneficial. The treatment for
patients with homozygous mutations is injections of
liraglutide, a GLP-1 receptor agonist that causes
weight loss by reducing appetite. The example of
obese people with the pathogenic MC4R mutation
treatment resulted in an average weight loss of 6.8 kg.
(Iepsen 2018) Overall fat, waist circumference,
fasting, and postprandial glucose concentration were
all decreased in a comparable way. As a result,
liraglutide might be an effective therapy for obesity's
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
216
most frequent homogenous mutation.
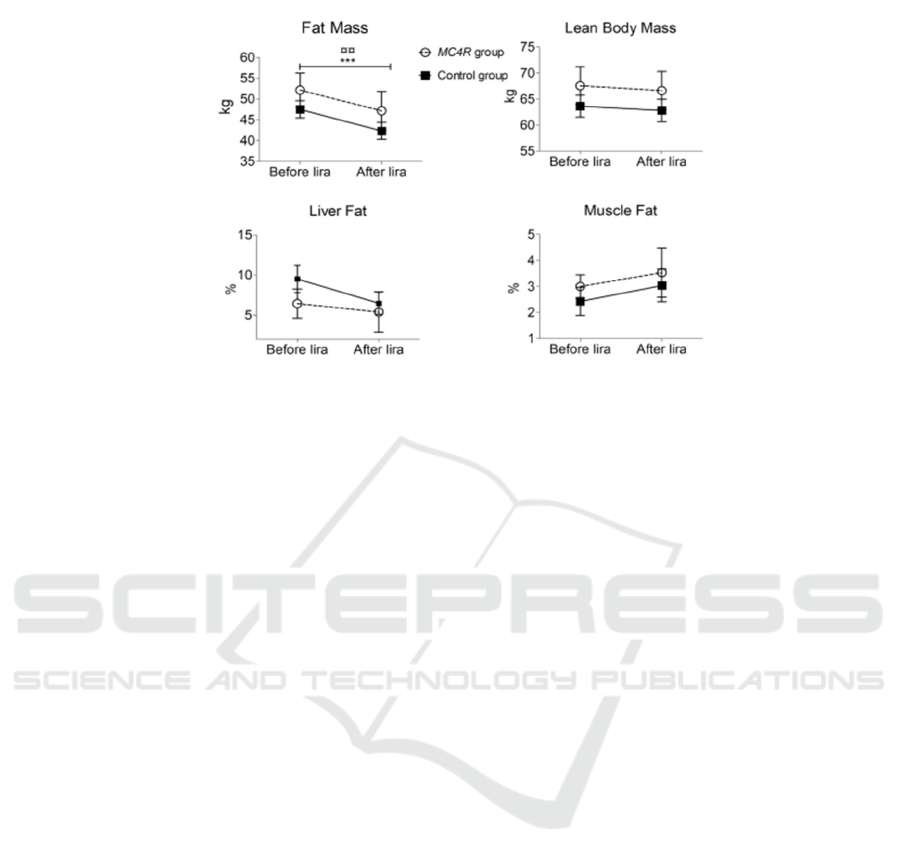
Figure 3: Results of liraglutide treatment for 14 people during 16 weeks (Iepsen 2018).
This graph is a comparison graph of 14 people
treated with liraglutide for 16 weeks done by Eva W.
Iepsen, Jinyi Zhang, et al. The graph shows the
comparison of fat mass, lean body mass, liver mass,
and muscle fat. There is a significant decrease in fat
mass in both MC4R deficient and controlled groups.
In addition, lean body mass and liver mass also
decreased. This fully demonstrates that liraglutide
treatment is helpful in MC4R deficient group. It is a
very good treatment option.
3.3
UCP Deficiency (Rosiglitazone)
UCP deficiency is not a direct cause of obesity in
people, but UCP deficiency can make that group of
people more susceptible to obesity. Moreover, UCP is
involved in the thermogenesis of brown fat which
helps burn fat. If UCP is deficient, the body will be
forced to use other thermogenic systems, which is one
less way to help the body burn fat and is another form
of obesity causing factor. Thus, the way to treat UCP
deficiency is to take rosiglitazone (Rosi), which is a
PPAR gamma agonist, usually in the form of a pill. It
can be taken with or without meals at regular times.
This drug was shown to be used in the treatment of
UCP because, in primary cultures of brown
adipocytes, rosiglitazone has an "exogenous" effect
and induces p44/p42 and p38 mitogen-activated
protein kinase (p38MAPK) activation. The latter is
involved in the expression of the UCP-1 gene. Thus,
the action of Rosi can lead to a high increase in
transcriptional activity on the UCP-1 enhancer,
leading to thermogenic effects. (Teruel 2002)
Ultimately it will help patients to stimulate
thermogenesis and fat oxidation in humans.
3.4
Beta 3-Adrenergic Receptors
Mutation (Mirabegron)
People with mutations in the beta3-adrenergic
receptor gene (Trp64Arg) typically develop non-
insulin-dependent diabetes with a low metabolic rate,
as well as increased body weight. Treatment is with
Mirabegron, a beta3-AR agonist that effectively
activates BAT. It will enhance glucose homeostasis
and treat several of the underlying mechanisms of
insulin resistance, such as insulin sensitivity and
secretion, while also improving adipose tissue and
inflammation. Mirabegron begins to work within a
few hours but may take several weeks to reach full
effect. (Finlin 2020)
3.5
PPARγ (Thiazolidinediones)
PPARγ is expressed predominantly in fat and very
low in the liver and muscle. PPARγ deficiency leads
to severe lipoatrophy, insulin resistance, and other
associated metabolic disorders. In addition to its
effects on obesity, PPARγ has a major impact on hair
formation, breast growth, and increased bone mass.
Thus, PPARγ is an important regulator of the
development and function of these adipose-
containing tissues. The treatment of PPARγ is with
thiazolidinediones (TZD). TZD is a synthetic ligand
for PPARγ. PPAR improves glucose and lipid
absorption, boosts glucose oxidation, decreases free
fatty acid concentrations, and reduces insulin
resistance via activating numerous genes in tissues.
Research on Obesity Caused by Genetic Defects in Specific Populations
217

Among the studied genetic defects that cause
obesity, congenital leptin deficiency is the least
frequent one, and it occurs in childhood. MC4R is the
most common genetic defect that causes obesity, and
its treatment is more straightforward than other
genes. One method is bariatric surgery, and the other
is liraglutide injections, which act as an appetite
suppressant. UCP deficiency does not lead to direct
obesity. Still, the study of mice have shown that UCP
deficiency can lead to a higher risk of future obesity
than in the general population. Both β3-AR and
PPARγ genes are critical, and deficiency is associated
with obesity and metabolic problems. In particular
the master gene PPARγ can cause problems in many-
body functions. β3-AR is treated with Mirabegron.
Mirabegron is an agonist of β3-AR itself, and PPARγ
is somewhat similar to β3-AR, but is treated with
Thiazolidinedione, which is a PPARγ agonist, is also
used to stimulate PPARγ to treat obesity. Although
TZD is beneficial for obesity and insulin resistance,
some studies have shown that the use of TZD, the
synthetic ligand of PPARγ, leads to bone-crunching,
which allows for higher fracture rates. Rosiglitazone
inhibits osteoblast differentiation and activates
osteoclast differentiation, leading to bone loss due to
reduced bone formation and increased bone
resorption. (Wei 2011) Therefore, more researches
are needed to find better solutions to the problem of
PPARγ deficiency.
4 CONCLUSIONS
This paper mainly focuses on the causes and possible
solutions of obesity caused by genes and metabolism
in populations with different genetic defects. And
also, this study has successfully summarized and
concluded the effects of each gene congenital or
mutation and the corresponding treatment options.
However, during the research process, it was found
that some genes, such as the UCP1 gene defect, do
not respond significantly in human bodies but are
inferred from experiments in mice, and further
clinical studies are needed in the future. Also, most of
the treatments found in this study were
pharmacological, and the PPARγ treatment drug
Thiazolidinediones had significant side effects,
which needs to be further investigated in the future
treatment of PPARγ gene defects.
REFERENCES
Abdullah, S., Reginold, W., Kiss, C., Harrison, K. J., &
MacKenzie, J. J. (2016, September 21). Melanocortin-
4 receptor deficiency phenotype with an interstitial 18q
deletion: A case report of severe childhood obesity and
tall stature. Case Reports in Pediatrics. Volume 2016, 6
Abdullah, S., Reginold, W., Kiss, C., Harrison, K. J., &
MacKenzie, J. J. (2016, September 21). Melanocortin-
4 receptor deficiency phenotype with an interstitial 18q
deletion: A case report of severe childhood obesity and
tall stature. Case Reports in Pediatrics. Volume 2016, 6
Chen, Y., Zhang, F., Heiman, M., & Dimarchi, R. (2005).
Leptin: Structure, function and biology. Vitamins and
hormones. volume71:345-372.
Chng, W. K. (2012, January). An overview of mongenic
and syndromic obesities in humans. Pediatric blood &
cancer. 58(1): 122–128.
Chuck, H. (2021, March 25). Leptin the appetite suppressor
hormone. Walking Off Pounds. Retrieved October 27,
2021, from http://walkingoffpounds.com/leptin-the-
appetite-suppressor-hormone/.
Clément, K., Al., E., Author AffiliationsFrom the Centre
National de la Recherche Scientifique, Others, A. J.
and, Others, C. A. B. and, Others, M. E. and, Others, J.
K. and, Others, N. B. and, & J. Lopez Bernal and
Others. (1970, September 2). Genetic variation in the
β3-adrenergic receptor and an increased capacity to
gain weight in patients with morbid obesity: Nejm.
New England Journal of Medicine. 333,352-354
Doulla, M., McIntyre, A. D., Hegele, R. A., & Gallego, P.
H. (2014, December). A novel MC4R mutation
associated with childhood-onset obesity: A case report.
Paediatrics & child health. 19(10): 515–518.
Finlin, B. S., Memetimin, H., Zhu, B., Confides, A. L.,
Vekaria, H. J., Khouli, R. H. E., Johnson, Z. R.,
Westgate, P. M., Chen, J., Morris, A. J., Sullivan, P. G.,
Dupont-Versteegden, E. E., & Kern, P. A. (2020, May
1). The β3-adrenergic receptor agonist mirabegron
improves glucose homeostasis in obese humans. The
Journal of Clinical Investigation. 130(5):2319-2331
Gao. M., Liu. D. (2014) Gene Therapy for Obesity:
Progress and Prospects,
https://www.discoverymedicine.com/Mingming-
Gao/2014/06/gene-therapy-for-obesity-progress-and-
prospects/
Kontani, Y., Wang, Y., Kimura, K., Inokuma, K.-I., Saito,
M., Suzuki-Miura, T., Wang, Z., Sato, Y., Mori, N., &
Yamashita, H. (2005, May 9). Ucp1 deficiency
increases susceptibility to diet‐induced obesity with
age. Wiley Online Library. Aging Cell 4, 147–155
Kozak, L. P., & Anunciado-Koza, R. (2008, December).
Ucp1: Its involvement and utility in obesity.
International journal of obesity (2005). 32(Suppl 7):
S32–S38.
Lazar, M. A. (2004, November 18). PPARγ, 10 years later.
Biochimie. 87: 9–13
Licinio, J., Caglayan, S., Ozata, M., Yildiz, B. O., de
Miranda, P. B., O'Kirwan, F., Whitby, R., Liang, L.,
Cohen, P., Bhasin, S., Krauss, R. M., Veldhuis, J. D.,
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
218

Wagner, A. J., DePaoli, A. M., McCann, S. M., &
Wong, M.-L. (2004, March 30). Phenotypic effects of
leptin replacement on morbid obesity, diabetes
mellitus, hypogonadism, and behavior in leptin-
deficient adults. Proceedings of the National Academy
of Sciences of the United States of America. Retrieved
October 28, 2021, from
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3847
81/. 101(13): 4531–4536 doi:
10.1073/pnas.0308767101
Melanocortin 4 Receptor. (2021, August 23). Accessed
September 6, 2021. https://www.mc4r.org.uk/
Paz-Filho, G., Delibasi, T., Erol, H. K., Wong, M.-L., &
Licinio, J. (2009, November 4). Congenital leptin
deficiency and thyroid function. Thyroid research. 2:
11. doi: 10.1186/1756-6614-2-11
Paz-Filho, G., Mastronardi, C., Delibasi, T., Wong, M.-L.,
& Licinio, J. (2010, November). Congenital leptin
deficiency: Diagnosis and effects of leptin replacement
therapy. Arquivos brasileiros de endocrinologia e
metabologia. 54(8): 690–697.
Pravednikova, A. E., Shevchenko, S. Y., Kerchev, V. V.,
Skhirtladze, M. R., Larina, S. N., Kachaev, Z. M.,
Egorov, A. D., & Shidlovskii, Y. V. (2020, May 25).
Association of uncoupling Protein (ucp) gene
POLYMORPHISMS With CARDIOMETABOLIC
DISEASES. Molecular medicine (Cambridge,
Mass.). 26: 51. doi: 10.1186/s10020-020-00180-4
Razzoli, M., Emmett, M. J., Lazar, M. A., & Bartolomucci,
A. (2018, May 1). β‐Adrenergic receptors Control
brown adipose UCP‐1 tone and cold response without
affecting ITS circadian rhythmicity. Federation of
American Societies for Experimental Biology. Volume
32, 5640 - 5646
Stienstra, R., Duval, C., Müller, M., & Kersten, S. (2007).
Ppars, obesity, and inflammation. PPAR research.
Volume 2007: 95974. doi: 10.1155/2007/95974
Teruel, T., Hernandez, R., Benito, M., & Lorenzo, M. (n.d.).
Rosiglitazone and retinoic ACID induce uncoupling
PROTEIN-1 (UCP-1) in a p38 mitogen-activated
Protein kinase-dependent manner in fetal primary
brown adipocytes. The Journal of biological chemistry.
278(1):263-9. doi: 10.1074/jbc.M207200200.
W Iepsen, E., Zhang, J., S Thomsen, H., L Hansen, E.,
Hollensted, M., Madsbad, S., Hansen, T., J Holst, J.,
Holm, J.-C., & S Torekov, S. (n.d.). Patients with
obesity caused By Melanocortin-4 receptor mutations
can be treated with A Glucagon-like Peptide-1 receptor
agonist. Cell metabolism. 28(1):23-32. e3. doi:
10.1016/j.cmet.2018.05.008.
Wang, F., Mullican, S. E., DiSpirito, J. R., Peed, L. C., &
Lazar, M. A. (2013, November 12). Lipoatrophy and
severe metabolic disturbance in mice WITH fat-
specific deletion of pparγ. Proceedings of the National
Academy of Sciences of the United States of America.
110(46): 18656–18661. doi: 10.1155/2007/95974
Wei, W., & Wan, Y. (2011, October 29). Thiazolidinediones
on pparγ: The roles in bone remodeling. PPAR
Research. vol. 2011,9. doi.org/10.1155/2011/867180
Xu, H., Barnes, G. T., Yang, Q., Tan, G., Yang, D., Chou,
C. J., Sole, J., Nichols, A., Ross, J. S., Tartaglia, L. A.,
& Chen, H. (2003, December). Chronic inflammation
in fat plays a crucial role in the development of obesity-
related insulin resistance. The Journal of clinical
investigation. 112(12): 1821–1830. doi:
10.1172/JCI19451
Research on Obesity Caused by Genetic Defects in Specific Populations
219
