
Precision Genome Editing towards the Treatment of
Hemoglobinopathies
Huimeng Sun
a
Queen Margaret’s School, Duncan, British Columbia, Canada
Keywords: CRISPR-Cas9, Base Editing, Sickle Cell Anemia, β – Thalassemia, Delivery Methods, Clinical Trials.
Abstract: Hemoglobinopathies, including sickle cell disease and β – thalassemia, are genetic disorders that cause
people to suffer from anemia. Apart from the lifelong therapeutic methods, gene therapy has been
introduced in the last decades of research as an efficacious treatment option, supported by various types of
delivery methods. In this work, I review the precision genome editing towards the treatment of
hemoglobinopathies. With a brief cover of the disease pathology and genome editing tools, special focus has
been directed towards the potential editing sites and clinical trials in progress.
1 INTRODUCTION
1
According to the World Health Organization
(WHO), approximately 7% of the global population
are carriers of hemoglobinopathies, which are
divided into thalassemia syndromes and structural
hemoglobin variants. The most common
hemoglobinopathies are β-thalassemia and sickle
cell disease (SCD). Patients suffering from β-
thalassemia and sickle cell disease (SCD) show
mutated or low levels of β-globin chain production.
The mutated β-globin chains result in the
hemoglobin tetramer to polymerize, which causes
the red blood cells to create a “sickle-like” shape
leading to vaso-occlusive crises and tissue damages.
Allogeneic hematopoietic stem cell (HSC)
transplantation together with iron chelation is the
only approved curative treatment for severe genetic
blood disorders. However, this method is
constrained by the availability of human leukocyte
antigen matching donors and face the limitation of
graft rejection. Collectively, these genetic blood
disorders are recognized as one of the major health
challenges in the world. which results in low
flexibility and induces hemolytic anemia and
vascular occlusions.
Over the past decade, new technological
breakthroughs in genome editing have propelled the
potential to cure genetic disorders. A cutting-edge
a
https://orcid.org/0000-0001-8322-2845
technology adapted from the CRISPR (clustered
regularly interspaced short palindromic repeats)/Cas
(CRISPR-associated protein) bacterial immune
system has been established as a platform for
performing more efficient, accurate, and precise
genome editing. These fundamental technologies
have led to the creation of groundbreaking tools that
have the capability to transform biological research
and curing genetic diseases. Traditional CRISPR
editing employs the creation of double stranded
breaks (DSBs) by endonucleases, which
subsequently stimulates DNA repair mechanisms.
These numerous developments have accelerated the
exploration of various curative strategies for the
treatment of hemoglobinopathies. Genome editing
approaches that generate DNA DSBs can correct the
disease-causing mutation or induce higher levels of
fetal hemoglobin (HbF) expression. However,
nuclease-mediated DSB-induced cytotoxicity and
chromosomal rearrangement severly limit these
approaches as eventual treatments. Recently
developed DSB-free strategies have overcome these
issues while maintaining superior clinical outcomes.
There are now many parallel clinical trials that
explore multiple editing strategies for the treatment
of β–hemoglobinopathies. Clinical studies using
lentiviral-based gene modifications have proved to
be successful at ameliorating the clinical symptoms
in patients with hemoglobinopathies. The
transplantation of genetically modified autologous
hematopoietic stem cells (HSCs) prevents any
immunological risks, such as graft reject. The
Sun, H.
Precision Genome Editing towards the Treatment of Hemoglobinopathies.
DOI: 10.5220/0011377400003443
In Proceedings of the 4th International Conference on Biomedical Engineering and Bioinformatics (ICBEB 2022), pages 1065-1075
ISBN: 978-989-758-595-1
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
1065

majority of the patients receiving edited HSCs
demonstrate reduced transfusion requirements while
maintaining high levels of fetal hemoglobin
expression. In this review, I present a comprehensive
overview comparing different types of genome
editing technologies and approaches for the
treatment of β–hemoglobinopathies.
2 OVERVIEW OF DISEASE
PATHOLOGY (SICKLE CELL,
BETA-THALASSEMIA, ETC.)
Sickle Cell Disease (SCD) is an autosomal recessive
genetic disorder affecting the function of
hemoglobin and was first discovered in 1910 by
James Bryan Herrick. Hemoglobin is tetrameric
molecule that formed by a globin group surrounded
by four heme groups and function as carriers of
oxygen (O2), carbon dioxide (CO2), and nitric oxide
(NO). They are critical in transferring oxygen (O2)
throughout the bloodstream from lungs to tissues
and cells, which is a major distinction of mammal
life. Sickle cell anemia is caused by an inherited,
single missense mutation in the β globin chain. The
single adenine to thymine base substitution results in
an amino acid conversion from glutamine to valine
in the globin gene. Normal red blood cells are
shaped like a binocave discoid, providing elasticity
to the cell and allowing red blood cells to pass
through the capillary bed to deliver oxygen.
However, the deoxygenation of sickle hemoglobin
(Hb S) results in an abnormally shaped erythrocyte
with a “sickling” appearance. This results in a
polymerization of sickle red blood cell which results
in low flexibility and induces hemolytic anemia and
vascular occlusions.
The β–thalassemia and α–thalassemia are two
major categories of thalassemia syndromes, in which
the patient lacks the production of β-globin chain. α–
thalassemias are a result of defects in the α–globin
chain of adult hemoglobin, including partial (α
+
) and
complete (α
0
) deletions or mutations of the four α–
globin genes. α –thalassemia can be further
categorized into 4 types based on different
genotypes and symptoms. α thalassemia silent
carriers are when only one gene is deleted or
damaged out of three; because only a single copy is
affected, the carrier does not show any clinical
symptoms nor a reduction in Hb values, but the
affected gene is inheritable through generations. α
thalassemina carriers have two missing genes that
result in mild anemia. Hemoglobin H disease is
caused by three missing genes that may lead to
moderate and severe microcytic hydrochronic
anemia resulting in impaired hemoglobin
production, with symptoms of fatigue, exercise
intolerance, and enlarged spleens. α - thalassemia
major is a fatal disease without treatment, in which
all four genes are missing that cause severe anemia.
β - thalassemia are caused by the insufficient (β
+
) or
absent (β
0
) production of β-globin chains in
hemoglobin and is highly prevalent in
Mediterranean countries. β- thalassemia can be
further divided into 3 types. Carriers of β–
thalassemia minor are normally clinically
asymptomatic. β- thalassemia intermediate cases
cause milder anemia and the majority of patients do
not require any blood transfusions to reduce chronic
anemia due to a compensation mechanism of
hypertrophy by erythroid marrow. β- thalassemia
major corresponds to symptoms of growth
retardation, progressive enlargement of abdomen,
and skeletal deformities. Although regular
transfusion medications are available, they may
result in problems of iron overloading which cause
dilated myocardiopathy, fibrosis, diabetes, etc.
Cardiac diseases are the most life-threatening
complication in patients treated with blood
transfusions.
3 OVERVIEW OF GENOME
EDITING (ZNF, TALEN, CAS9,
BASE EDITING, PRIME
EDITING, AND MORE)
Over the decades, the emergence of gene editing
technologies has offered scientists the ability to
introduce modifications to the genome of various
cell types in hopes of creating genetic therapeutics.
The use of highly specific and programmable
nucleases that introduce double-stranded breaks
(DSBs) provides the ability to specifically edit any
the region of interest. DSBs are repaired by
endogenous cellular mechanisms, either through a
non-homologous end-joining (NHEJ) repair pathway
that generates insertion and deletions (indel), or
homology-directed repair (HDR) pathway that
utilizes a genetic donor template to replace the DNA
surrounding the DSB. Meganucleases (homing
endonucleases), the earliest gene technology, are
sequence-specific DNA cleavage enzymes used in
targeting, replacing, and modifying the genome.
Meganucleases, exist as dimers or single-chain
enzymes, bind and cleave sequences that are at least
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1066

12 base pairs in length with high specificities.
Although effective at manipulating DNA,
meganucleases have one major limitation. The
biggest limitation is that for a meganucleases to bind
to DNA, the protein requires specific sequences
upon which only this protein can bind to the
matched DNA sequence. If one wishes to change the
target DNA sequence, a whole different protein, or a
profound protein engineering effort of the
meganucleases is needed to evolve and re-engineer
the protein, which makes it difficult for widespread
usage of these tools across biomedical research and
biomedicine.
The next two big classes of gene editing
technologies developed are zinc finger nucleases
(ZFN) and transcription activator-like effector
nucleases (TALEN). ZFNs consist of engineered
zinc fingers fused to a non-specific, dimeric FokI
nuclease domain. DNA-contacting residues can be
replaced by connecting individual zinc fingers with
novel DNA-binding specificities. However, there is
a grand challenge when making multi-finger arrays
as the fingers are not always modular. Each
individual zinc finger recognizes a specific 3-base
pair sequence, but each finger also has context-
dependent effects, so it is not always as simple as
replacing the finger with the desired targeting
sequence. TALENs are modular DNA-binding
protein units derived from naturally occurring
TALEs (from Xanthomonas). Each monomeric
TALE repeat recognizes a specific type of DNA
base. Like zinc finger arrays, individual TALE
subunits can be simply joined together to recognize
longer sequences of DNA. Similar to ZFNs, TALE
repeat arrays can be fused to a dimeric FokI nuclease
domain to create TALENs. Several differences are
noted between ZFNs and TALENs. The biggest
distinction between them is that each monomeric
TALE subunit binds to only a single nucleotide
instead of a nucleotide triplet, which makes TALE
targeting simpler than that of ZFNs. From a cost and
time perspective, TALENs are cheaper and faster to
produce, and are more flexible and easier to design
because of their simple DNA recognition properties
compared to ZFNs. Immunogenicity is another huge
consideration for using ZFNs and TALENs in
therapeutic applications. ZFNs display little to no
immunogenicity because the sequences are found in
all organisms from yeast to humans, while the
immunogenicity for TALENs in humans is still
unknown. However, it is predicted that the immune
response of TALENs may be relatively higher since
the sequences of TALE repeats are only found in
Xanthomonas plant pathogens.
Although meganucleases, ZFNs, and TALENs
all have their distinct advantages, there are still
several limitations remaining to their widespread.
The primary difficulty in using these technologies is
that they are fundamentally dictated by a protein-to-
DNA interaction when driving specificity of DNA
targeting. Therefore, an extensive protein
engineering and optimization effort is needed when
one wants to target a new sequence of DNA.
Furthermore, the cost and time needed to produce
each variant is a huge limitation that needs to be
considered. It takes weeks to months to produce one
specific agent that then still needs to be optimized
for a specific site, and many times prices go beyond
the financial capability of people and researchers
with goals of routine usage. Therefore, there remains
a need for an alternative approach that can overcome
the remaining limitations in genome editing.
Clustered regularly interspaced short
palindromic repeats (CRISPR) and CRISPR-
associated protein 9 (Cas9) is the latest technology
that revolutionized the field of genome editing.
CRISPR-Cas9 was first identified in 2008 as a
bacterial defense mechanism that host bacteria
leverage against bacteriophage infections in nature.
This was later repurposed as a gene editing tool that
uses Cas9 as a nuclease guided by a single guide
RNA sequence to bind to a specified target DNA
sequence. Although CRISPR-Cas9 is primarily
driven by RNA-DNA interactions, there is one
remaining protein-DNA interaction known as the
protospacer adjacent motif (PAM). The PAM
sequence is required for Cas9 binding to a target
genomic site. Following Cas9 binding, a small DNA
R-loop is formed and a subsequent DNA double-
stranded break is generated by the Cas9
endonuclease.
One of the critical aspects of genome editing to
evaluate is the off-target propensity of each editing
agent. The guide RNA in Cas9 dictates a specific 20
base pair sequence of the targeted DNA sequence.
However, Cas9 binding can tolerate small
mismatches between the guide RNA and target DNA
sequence, meaning that there are possible binding
sites that are not the specified genomic site. Each
one of these undesired binding sites are known as
off-target sites in genome, which may cause
undesirable and dangerous negative effects that
could lead to additional genetic diseases and
unwanted phenotypes, such as cancer. Engineered
high-fidelity Cas9 variants have been developed as
an effective approach to decrease off-target effects.
SpCas9-HF1 is considered to be a high-fidelity
variant that maintains high on-target DNA editing
Precision Genome Editing towards the Treatment of Hemoglobinopathies
1067

efficiency (at least 70% of the wild-type SpCas9),
but dramatically reduced DNA off-target editing
efficiencies. The route of delivery into the cell also
has a role in the propensity of off-target editing.
Protein delivery further reduces off-target effects
compared to DNA viral or plasmid delivery since
DNA produces transcripts with subsequently product
a plethora of editor proteins. Furthermore, the life
span of DNA is longer compared to both RNA and
protein, which contributes to a longer exposure of
editor protein in cells. Because each cell only
contains a limited amount of DNA substrate for the
editing event, any additional editor protein has the
propensity to result in off target editing. Therefore,
delivering genome editing protein directly can
significantly decrease off target effects.
There are a variety of strategies developed
towards detecting Cas9 off target sites, which is vital
towards increasing the efficacy and safety of
CRISPR-Cas9 genome editing systems. At a single
cell level, whole genome sequencing delivers a
comprehensive view of the entire genome at a DNA
base level. However, off-target effects are
ineffective and occur at a relatively low percentage,
therefore requiring tens, hundreds, or even
thousands of single cell genome sequencing
evaluations to thoroughly evaluate all off-target
events. Therefore, it is expensive and not effective to
rely upon whole genome sequencing to thoroughly
evaluate the off-target propensity of every genome
editing agent. Alternative approaches to whole
genome sequencing were introduced that can enrich
for off-target editing events. These rely
fundamentally upon next-generation sequencing
techniques. GUIDE-seq (Genome-wide, Unbiased
Identification of DSBs Enable by Sequencing) and
CIRCLE-seq (Circularization for In vitro Reporting
of CLeavage Effects by sequencing) are two
methods that enrich for Cas9 off-target sites that
experience a DSB. There are computational methods
that can predict potential off-target sites in the
genome, but these computational methods are
deemed a preliminary and biased compared to
unbiased experimental approaches. GUIDE-seq
integrates a dsODN (double-stranded
oligodeoxynucleotide) donor template into cleavages
sites by non-homologous end joining repair inside a
cell’s genome directly. However, NHEJ is prone to
produce indels rather than incorporating exogenous
sequences, which suggests that the sensitivity of
GUIDE-seq is one aspect that still needs to be
considered. CIRCLE-seq is a highly sensitive in
vitro assay that serves as an alternative approach to
GUIDE-seq in detecting off target sites. CIRCLE-
seq relies on first generating and purifying libraries
of circularized genomic DNA, then treating this pool
of circles with Cas9 nucleases. Subsequently,
nuclease-linearized DNA fragments are ligated with
sequencing adapters and sequenced using next
generation sequencing to identify genome-wide
Cas9 nuclease off target sites.
Base editing is a relatively new genome editing
technology that is able to edit DNA without any
DSB intermediate. Base editing enables the ability to
perform chemistry directly on the genome of living
cells. Base editing fundamentally relies upon the
Cas9 component from the CRISPR-Cas systems
fused together with an enzyme that directly modifies
DNA or RNA. Dead Cas9 (dCas9) or nickase Cas9
(nCas9) is first guided by a sgRNA to the targeted
region in the genome. Following Cas protein
binding, the Cas protein exposes a single-stranded
DNA R-loop region that serves as substrate for
deamination mediated by deaminases fused to the
Cas protein. Base editing is currently largely
grouped into either cytosine BEs that can convert
cytosine-guanine (C-G) to thymine-adenine (T-A)
base pairs, or adenine BEs that can convert adenine-
thymine (A-T) to guanine-cytosine (G-C) base pairs.
Base editors create precise, predictable, and efficient
genetic outcomes at the targeted sequence without
any undesired indel byproducts or large genomic
perturbations such as p53 activation, DNA
rearrangements, or DNA translocations.
Similar to base editing, prime editing is able to
perform gene editing without inducing DSBs. But
prime editing can generate a wider range of possible
alterations as it can perform all twelve possible base-
to-base conversions. Prime editing is composed of a
Cas9 nickase fused to PE2 (a modified reverse-
transcriptase) and a multifunctional prime editing
guide RNA (pregRNA). A major advantage of prime
editing is its low off-target activity compared to
Cas9 as edition of PE2 significantly lowered the off-
target effects with fewer indels. Moreover, PE
breaks the bottleneck in therapeutic application of
gene editing as HDR machinery is not required for
prime editing so that post-mitotic cells can be edited.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1068
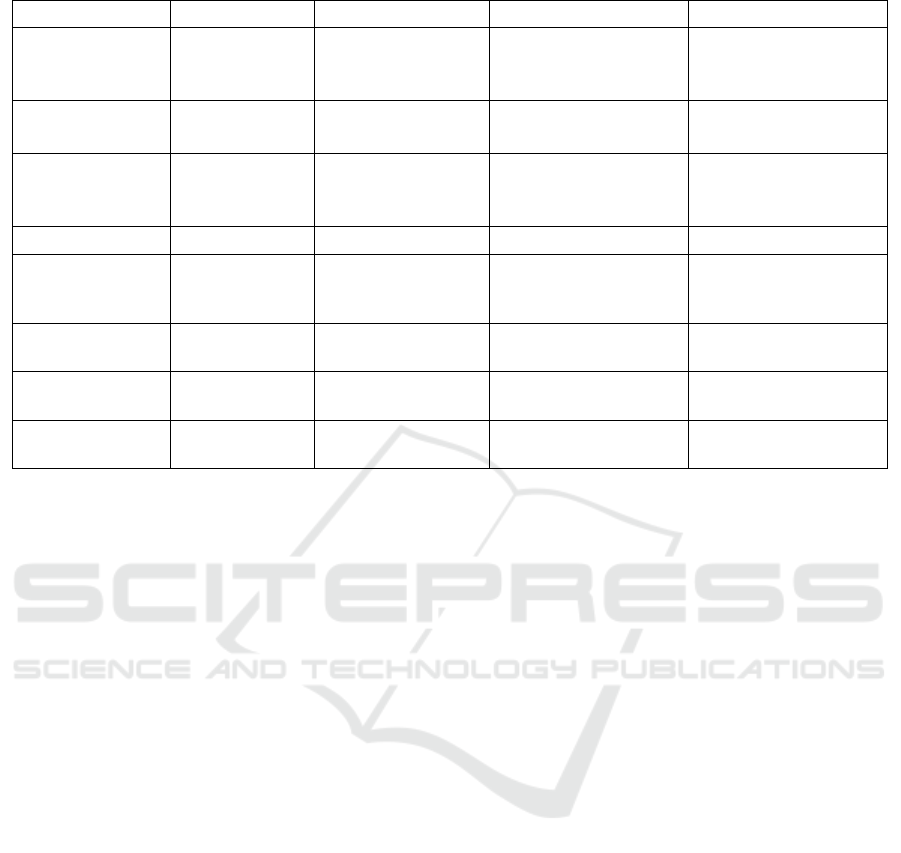
Table 1: Comparison of four commonly used genome editing biotechnologies.
Technology
Meganuclease ZFN TALEN CRISPR/Cas9
Origin
Microbial
genetic
elements
Eukaryotic gene
regulators
Bacterium
Xanthomonas
Adaptive immune
system in archaea and
bacteria
Targeting
Protein – DNA
interaction
Protein – DNA
interaction
Protein – DNA
interaction
RNA – DNA
interaction
Specifi
cDNA binding
elements
Tripletconfined zinc
finger proteins
Single-base
recoognition TALE
proteins
sgRNA
Off-target Effect
Low High Moderate Variable
Delivery
Major vector
systems
Major vector
systems
DNA, mRNA,
adenovirus, AAV
DNA, mRNA, viral
vectors with sufficient
packaging capacity
Time
Long (7-15 days)
Relatively long (5-7
days)
Short (1-3 days)
Targeting
Efficiency
Low Moderate Moderate High
Size
1kb*2 ~3kb*2
4.2kb (Cas9) +
1kb(RNA)
4 INITIAL USES OF DNA DSB
CUTTING FOR TREATMENT
Bone marrow and stem cell transplants are currently
the primary route of treatment for sickle cell disease
and β-thalassemia. In the last decade, the profound
discovery and development of genome editing
technologies has enabled the ability to use nucleases
that create a DNA double stranded breaks (DSBs) in
a specific region of the genome to be considered as
an initial and effective treatment of β-
hemoglobinopathies.
Genes that encode for the β-globin and α-globin
are located on chromosomes 11 and 16 in the human
genome, respectively. Embryonic hemoglobin, fetal
hemoglobin, and adult hemoglobin are three variants
of the hemoglobin protein expressed on the
erythrocyte at different times during development.
Fetal hemoglobin (HbF) is a tetramer consisting of
two α-globin chains and two β- globin chains.
Following the first three months post-conception, the
embryonic globin experiences a dramatic decrease in
globin synthesis, while the level of fetal hemoglobin
rises rapidly, serving as the primary form of
hemoglobin expressed in the fetus while in utero.
After the infant is delivered, the level of fetal
hemoglobin drops significantly, while the expression
of adult hemoglobin becomes the dominant form of
hemoglobin.
A regulatory region named as the locus control
region, LCR, regulates the expression of the encoded
globin genes, and is located 40 to 60 Kb upstream of
the beta-globin locus. BCL11A is a protein
expressed on the LCR region that binds to two
regions on the hemoglobin locus, HBG2 and HBG1,
which represses fetal hemoglobin expression,
signaling the production of adult globin genes. As
the mutation affecting sickle cell is located in the
adult globin gene, many therapeutic options being
explored using genome editing rely upon
upregulation and reactivation of fetal hemoglobin
expression to compensate for the sickled adult
globin gene. A reasonable approach to elevate the
amount of fetal hemoglobin expressed is to reduce
the BCL11A repressor activity. Certain mutations
found in the erythroid enhancer region can naturally
repress the amount of BCL11A being translated,
which ameliorates disease symptoms through fetal
hemoglobin compensation. Increased expression of
fetal hemoglobin is a sufficient at alleviating
symptoms of sickle cell because the fetal
hemoglobin has a similar to higher oxygen
saturation compared to that of adult hemoglobin. It
is encouraging that ongoing clinical trials have also
demonstrated that the induction of HbF can
ameliorate symptoms of sickle cell disease and β-
thalassemia.
Precision Genome Editing towards the Treatment of Hemoglobinopathies
1069
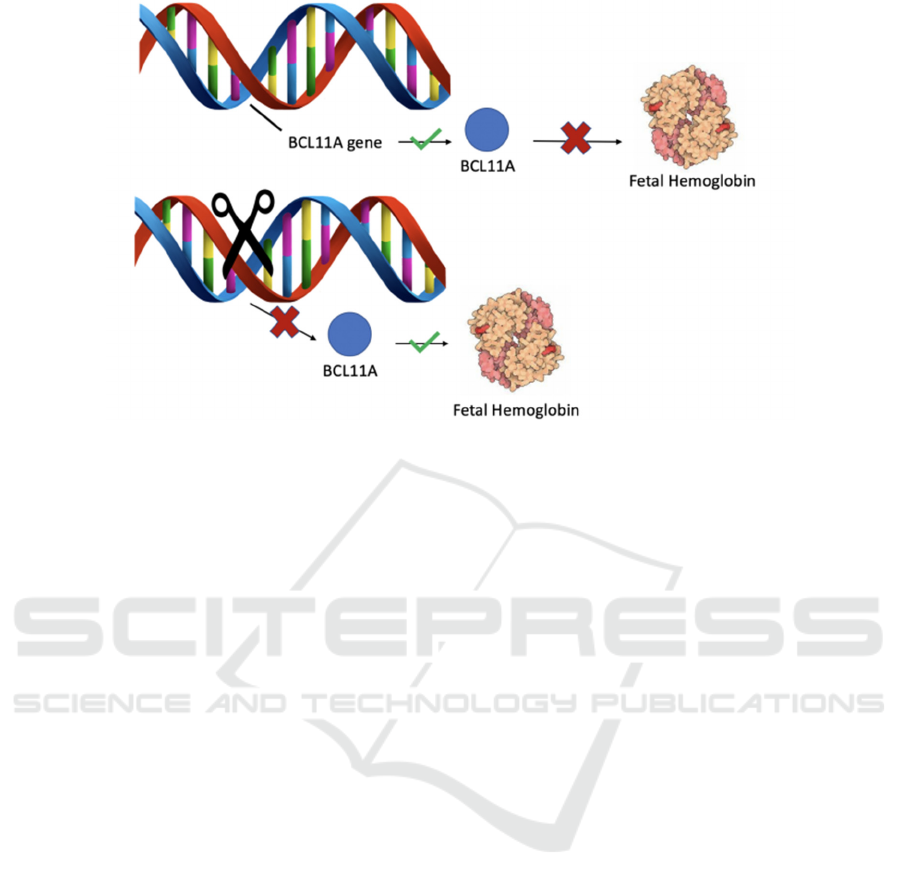
Figure 1: Visual Representation of the Mechanism of Deleting the BCL11A gene.
CRISPR-Cas9 gene editing is helping to tackle
sickle-cell disease in many ways. Firstly, Cas9
enzyme can be introduced with a guide RNA to
target and repair the faulty β-globin gene through
HDR replacement using an exogenous donor
sequence as the template to correct the disease-
causing mutation. The correction rate ranges from
7% to 50% depending on the editing tool used, such
as ZFNs, TALENs, and CRISPR/Cas9, and the
delivery method employed when delivering these
tools into induced pluripotent stem cell (iPSCs) and
HSPCs in vitro. However, xenotransplantation
experiments revealed that HDR lacks the generation
of long-term engraftable HSCs, shown by a 10%
drop in gene correction in vivo. This is a major
limitation to this direct replacement strategy so other
approaches are needed. Alternatively, a Cas9
enzyme can delete the gene encoding the BCL11A
repressors to increase the production of HbF. Like
previously discussed, fetal hemoglobin serves as a
sufficient replacement of adult globin in alleviating
sickle cell disease symptoms. Although these
approaches have been demonstrated to be effective,
the biggest drawback to using CRISPR-Cas9
nuclease is the creation of double-stranded breaks.
These DSBs have the potential to result in large
cellular perturbations such as chromathripsis,
activation of p53 pathways, and increased cell stress
signaling. Therefore, there is an urgent need for
alternative approaches of using genome editing
without any double-strand DNA break
intermediates.
5 PRECISION EDITING USING
BASE EDITING, PRIME
EDITING
Contrary to CRISPR-Cas9 nuclease editing, base
editing can generate direct conversion of one base
pair to another at a target site without any double-
stranded break intermediates. The DSB-free base
and prime editing systems ensure a superior safety
aspect of genetic modifications related to the
therapeutic treatment for β -hemoglobinopathies.
Two major classes of base editing currently exist: A-
to-G base editor (ABE) and C-to-T base editor
(CBE) that both are mediated by a deamination
reaction of either adenine or cytosine, respectively.
Base editing has the potential to be one of the most
superior gene editing technologies.
Two approaches employing base editing are
being explored in treating sickle cell disease: fetal
hemoglobin activation and direct correction of the
sickle-causing mutation. The first approach relies
upon recreating a phenomenon known as hereditary
persistence of fetal hemoglobin (HPFH) with base
editing. HPFH is a benign genetic condition
reflective of high levels of HbF expression in adults,
caused by either large deletions or point mutations in
the globin locus.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1070
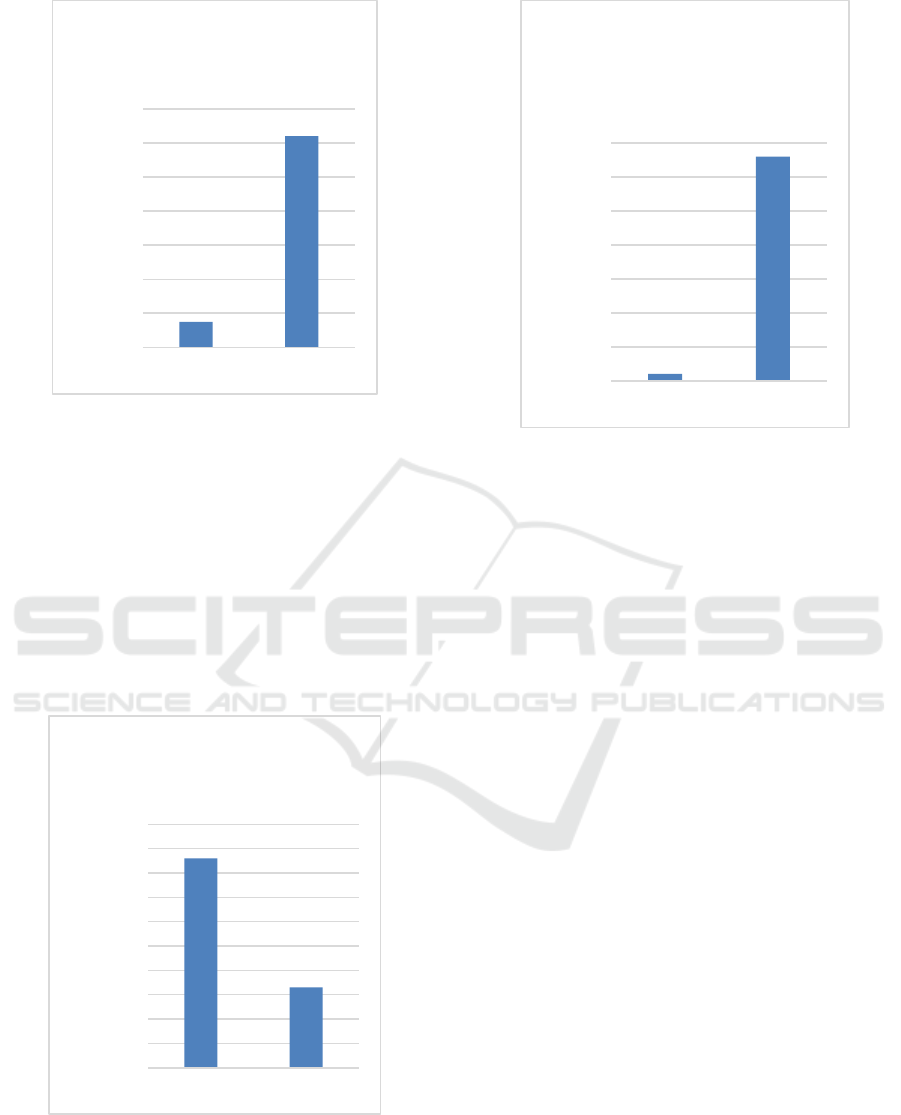
Figure 2: Levels of HbF y-globin protein detected in
unedited and edited erythroid cells.
ABE can induce mutations found in HPFH on
the fetal hemoglobin genes, HBG1/2, without
generating any double-stranded break intermediates
or undesired cellular perturbation side effects.
Significantly, over 80% base editing was achieved at
the HBG1/2 promoters, and this translated to a 60%
increase in the expression of fetal hemoglobin in red
blood cells. Moreover, high levels of editing and
robust HbF induction are maintained after a long-
term in vivo engraftment study in mice.
Figure 3: Level of HbS sickle protein detected in unedited
and edited erythroid cells.
Figure 4: Level of y-globin protein levels in erythroid cells
after 16 weeks of editing.
After 16 weeks, 90% base editing at the HBG1/2
promotors was maintained in the erythroid and more
than 65% of the cells expressed fetal globin. The
second approach in using base editing for sickle cell
is the creation of the Makassar mutation. The
Makassar variant is a naturally occurring mutation at
the sickle site where beta globin has a glutamine
instead of alanine. This variant can be found in a
portion of the asymptomatic human population in
Northern Europe, suggesting that it is an alternative
beta globin genotype that maintains life symptom-
free. The Makassar mutation allows the β-globin to
function normally, which is an important discovery
because the sickle cell mutation requires a base edit
that converts adenine to thymine, a conversion from
purine to pyrimidine that is not currently possible
with existing technologies. The Makassar variant
relies upon an adenine to guanine mutation at the
sickle site, which can be achieved using an adenine
base editor. Experimental data showed that 80% of
sickle cells can be corrected into this Makassar
variant using ABE in cells from a sickle patient.
Studies demonstrate that the Makassar mutation is
successful in the elimination of the HbS globin. In
untreated controls, 100% of cells are classified as
sickle cells; in contrast, just 80% successful base
editing at the targeted region reduced the amount of
sickle cells to only about 10% of the total
population.
0
10
20
30
40
50
60
70
Unedited Edited
y-globin/total β-globin (%)
Levels of HbF gamma
globin protein
0
10
20
30
40
50
60
70
80
90
100
Unedited Edited
βs-globin/Total β -like globin (%)
Levels of HbS sickle
protein
0
10
20
30
40
50
60
70
Unedited Edited
y-globin/Total β-like globins (%)
y-globin protein
levels in erythroid
cells
Precision Genome Editing towards the Treatment of Hemoglobinopathies
1071

6 TARGET CELL TYPES AND
DELIVERY METHODS
The delivery of genome editing agents to desired
cell types is a grand challenge achieving precise and
effective gene editing. The molecular weight of
Cas9 protein 160kDa and the phosphate backbone of
the sgRNA creates an overall negative charge to the
Cas9 complex. There are two main pathways being
employed for Cas9 delivery: in vivo strategies and
ex vivo strategies. Each of these two approaches has
its unique advantages and disadvantages so they
each can be used in different circumstances.
Electroporation is a physical delivery method being
explored ex vivo. Electrical currents are used to
stimulate cells to create an instantaneous opening of
pores in the cell membrane to make it permeable and
enable the delivery of surrounding substrates.
Electroporation is widely used in ex vivo gene
editing since it is capable of being applied to a wide
range of cell types. However, the electrical current
generated by electroporation results in a high
percentage of cell death so it is difficult to scale this
method for widespread adaptation.
Lipid delivery is another commonly used
delivery approach relying upon the formation of
lipid nanoparticles to encapsulate substrate cargo.
Lipids are comprised of a hydrophilic polar head and
a long hydrophobic nonpolar tail. The encapsulation
of negatively charged nucleic acids into positively
charged liposomes aids in the entry of these
nanoparticles into cells through cellular endocytosis.
Once in the cell, the nanoparticle is degraded upon
traveling to late endosomes, which releases the
cargo and permits downstream genome editing.
Previous efforts have demonstrated that these lipid
nanoparticles can effectively package DNA, RNA,
and ribonucleoprotein complexes. Furthermore,
studies have demonstrated that direct injection of
these nanoparticles enable effective delivery into the
liver of living animals.
Viral vectors are another effective approach
being explored for gene delivery due to the
ubiquitous nature of viruses in the world. Most
commonly used viruses in research include the
adeno-associated viruses (AAVs), lentivirus, and
adenovirus. AAVs serve as a main vector to deliver
Cas9 to differentiated tissues by transduction.
Several clinical trials have been approved using
AAVs, which are less immunogenic compared with
other viruses. Although AAVs are effective at
delivery transgenes into cells in animals and
humans, a grand challenge for AAVs is the
limitation on the carrying capacity. AAV vectors are
typically limited to genes smaller than 4.8 kb, so any
gene larger would be unable to be delivered using
one AAV vector. The delivery of Streptococcus
pyogenes Cas9 (SpCas9) by AAVs is challenging
due to its large size of 4.2 kb, while the delivery of
Staphylococcus aureus Cas9 (SaCas9) is a more
feasible approach with a size of 3.15 kb. In addition
to the Cas protein, one would also need to package
the sgRNA so it is difficult to package everyone
onto one AAV vector. Previous studies have used
two AAVs to deliver an SpCas9 (4.8kb) and a
sgRNA (3.0kb) separately into a mouse brain to
target a gene called Mecp2. Furthermore, they
demonstrated that they could include two additional
sgRNAs into the virus and obtain multiple edits in a
single cell. In a subsequent study, they describe that
SaCas9, about 1kb smaller than SpCas9, and its
sgRNA can fit one a single AAV vector with a size
of 4.7kb and demonstrate that this single AAV
vector can be delivered into mice liver in vivo.
Lentivirus (LV) is another type of viral vector
used to deliver CRISPR-Cas9 ex vivo. LV vectors
have a more generous packaging capacity of 8kb,
which allows for including both a Cas9 protein and a
targeting sgRNA into a single LV vector. Lentiviral
vectors are mainly used in ex vivo gene delivery and
are currently used as the delivery vector in FDA-
approved chimeric antigen receptor-T therapies.
These approaches have shown superior delivery
efficacy in hematopoietic stem cells and T cells.
However, the limitation of the scope of targets for
LV is larger than AAV, and previous studies have
limited any in vivo delivery prospects of using LVs
in preclinical trials. Another challenge to using LVs
is the genotoxicity and immunogenicity defects.
However, integrase-defective lentiviral vectors
(IDLVs) have been introduced to allow for efficient
and continual transgene expression in vivo while
minimizing any undesired cellular effects.
Adenoviruses (AdVs) are widely used in clinical
trials for gene delivery as AVs are able to transduce
both dividing and nondividing cells. The genome of
AdVs generally range from 34 – 43 kb long and
AdVs do not integrate into host cell genomes, which
minimizes any potential off-target effects. However,
a major concern of using AdVs as a delivery method
is that AdVs trigger intense immune responses,
which leads to significant inflammation.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1072
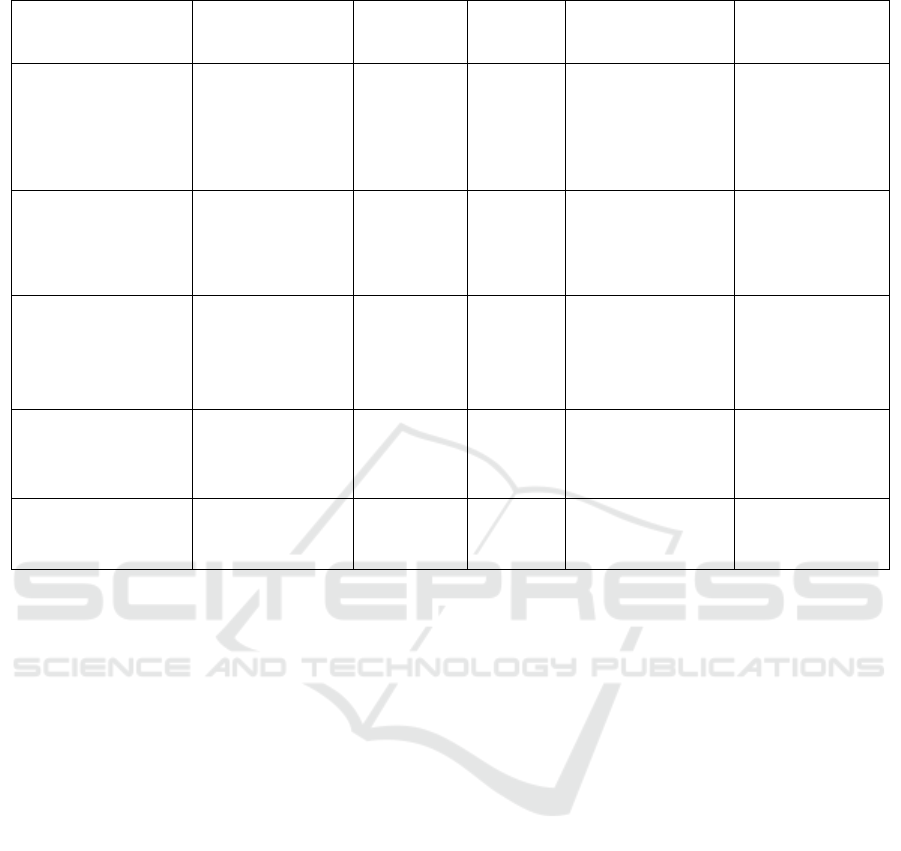
Table 2: Delivery Method Comparison Chart.
Method Delivery Material Apprpach
Packaging
Capacity
Advantages
Disadvantages
Adenoviruses
Double-stranded
DNA
In vivo 8-30kb
1.High transfection
efficienc
2.Transduce both
dividing and non-
diving cells
High
immunogenicity
Adeno- associated
viruses
Single-stranded
DNA
In vivo <4.8kb
1.Low
immunogenicity
2.High
transfection
efficienc
y
Limited
packaging
capacity
Lentiviruses
Single-stranded
RNA
In vivo 8kb
1.High transfection
efficienc
2.Decent
packaging capacity
Potential
genotoxicity
Electroporation
DNA plasmid,
mRNA, RNP
Ex vivo -
1.Viralfree2.Fast
3High transfection
efficiency
Lowcell viability
Expensive
Lipid Nanoparticles
DNA plasmid,
mRNA, RNP
Ex vivo -
1.Viral free
2.Cheap
High toxicity
A major challenge to using viruses to deliver
genome editing agents is that many newer precision
genomes editing approaches are larger than the
original Cas9 system. For instance, if using base
editing, the fusion of a deaminase with Cas9
enlarges the size of the overall editor complex. A
recent study demonstrate that it is possible to split
the base editor in half and linking them together
using an intein system. Each half could be packaged
into two separate AAV systems and can reconstitute
into a full-length base editor upon delivery into a
target cell. This system was used to demonstrate
successful base editing in a mouse brain, and as a
result, in both cortex and cerebellum, about 50% of
base editing was observed in the targeted cells.
Although most efforts for treating
β−globinopathies rely upon ex vivo approaches, a
recent study explored the possibility of in vivo
genome editing. It is known that editing the
autologous HSCs demonstrate a prolonged benefit in
treating SCD. Ex vivo delivery of genome editing
agents for hemoglobinopathies begin by taking out
the stem cells of patients to perform gene editing in
the lab, and then reintroduce the edited stem cells
back to the patient. A significant limitation is when
delivering the edited cells back to the patient, the
patients need undergo myeloablation, which is a
process of significantly weakening the natural
immune response edited stem cells can survive and
engraft in the patient. This process in patients can
generate other types of detrimental disorders and be
very damaging to the patient’s overall health. In new
data released by Intellia Therapeutics, they
demonstrate that certain LNPs can deliver CRISPR-
Cas9 mRNA into hematopoietic cells direct in vivo.
Lipid nanoparticle with the Cas9 mRNA and the
target gRNA were delivered into the cells, and they
observed editing in the stem cells of animals treated
with these engineered LNPs. Increased editing was
observed with repeat multi-dosing. Intellia achieved
therapeutic levels of editing in human CD34+ cells
in a xenotransplanted mouse. Overall, this data
suggests that using engineered LNPs is a safe and
effective process to deliver genome editing agents
directly into animals with minimal side effect.
7 DISCUSS CLINICAL TRIALS IN
PROGRESS
There are many approaches in which genome editing
is being explored for the treatment of
hemoglobinopathies. In this final section, I will
Precision Genome Editing towards the Treatment of Hemoglobinopathies
1073

discuss ongoing clinical trials conducted by Editas
Medicine, Beam Therapeutics, CRISPR
Therapeutics and Vertex Pharmaceuticals.
CRISPR-Cas9 mediated nuclease genome editing
through double-strand break intermediates is
considered as an initial and effective approach to
treat hemoglobinopathies. EDIT-301, introduced by
EDITAS, is an approach of editing HBG1/2 to
increase fetal hemoglobin expression as a
compensatory mechanism for sickled adult globin.
Key regulatory regions in the β-globin locus are
shown to be edited by SpCas9 and Cas12a. They
demonstrate that Cas9 and Cas12a editing allows for
a durable maintenance of indels at the target site.
After editing the HBG1/2 region, they demonstrate
that there is a constant amount of erythroid and
caspases produced, and there is no significant
increase in cell death. They evaluated that stem
blood cells treated with HBG1/2 editing displayed a
52% increase in the expression of fetal hemoglobin,
and that 89% of red blood cells will carry the fetal
hemoglobin compared to only 4% of red blood cells
when unedited.
Beam Therapeutics is a biotechnology company
that uses base editing as a therapeutic approach to
treat patients suffering from serious diseases. Beam
Therapeutics is exploring two uses of base editing
towards the treatment of hemoglobinopathies. First,
they use base editors to induce single base changes
in the regulatory regions of HBG1 and HBG2 to
disrupt repressor binding binds, which results in an
increased expression of fetal hemoglobin (HbF).
Second, they are exploring the use of adenine base
editing to directly edit the adenine implicated in
sickle cell disease to correct the E6V mutation into a
glutamic acid to reflect the Makassar variant. Their
initial data demonstrates that the Makassar program
is able to achieve 0% to 70% direct editing of the
sickle cell point mutation, which is sufficient
towards the alleviation of sickle cell symptoms.
A collaborative clinical trial between CRISPR
Therapeutics and Vertex Pharmaceuticals has
released preliminary findings on their therapeutic
program, CTX001, that targets BCL11A to result in
increased fetal hemoglobin expression. They
demonstrate that by editing patients’ own blood stem
cells with CRISPR-Cas9, they can achieve elevated
levels of HbF in red blood cells. The most recent
data reflect that five patients with beta thalassemia
and two patients with sickle cell disease treated
under CTX001 all have experienced successful
engraftment of edited blood stem cells and that they
had no vaso-occlusive crises (VOCs) during the
follow-up after the CTX001 infusion. Importantly,
all patients maintained near-normal hemoglobin
levels and showed drastic alleviation of
hemoglobinopathy symptoms.
It is exciting to continue witnessing the rapid
development of genome editing towards the
treatment of detrimental human disorders.
8 CONCLUSIONS
The rapid development of new genetic therapies
support a prosperous future for curing intractable
genetic disease. In the last few years, we have
witnessed the development of new technologies,
experimental models, and pre-clinical and clinical
studies. Insights into mechanism of action
demonstrated many possibilities of editing the β–
globin gene to introduce effective therapeutic
strategies that treat β–hemoglobinopathies.
While many encouraging clinical trials have been
released, many challenges still exist until we can
fully appreciate the full potential of these approaches
for curing these blood disorders. Firstly, the off-
target effects on DNA can potentially cause
irreversible damages, such as large genomic
rearrangements. Thus, there is a need to carefully
monitor these undesired events in clinical trials to
precisely plan for and adjust for any detected effects.
Specifically, the off-target activity of BEs, including
both sgRNA-independent and sgRNA-dependent
events require close monitoring. Fortunately,
engineering modifications into the deaminase have
lowered sgRNA-independent DNA off-target
activity while maintaining highly efficient on-target
DNA editing. Secondly, even though BE and PE
have overcome cytotoxicity events caused by DSBs-
induced indels, proper delivery methods are still
needed, especially for primary cells. However, it
remains a challenge to develop efficient methods
that can deliver the large complex sizes of BE and
PE technologies. Thirdly, the current pool of BEs
only enable C-T, C-G, and A-G conversions;
therefore, more optimizations and technologies are
required to enable other types of conversions. Prime
editing is a new approach that can overcome many
of the editing types possible; however, further
optimizations are needed to realize higher editing
outcomes. Despite these challenges, gene and cell
therapy hold great promise for providing proper
treatment approaches for patients diagnosed with β–
hemoglobinopathies.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1074

ACKNOWLEDGEMENTS
If any, should be placed before the references
section without numbering.
REFERENCES
Ahmad M. Khalil. 2020. The genome editing revolution:
review. Journal of Genetic Engineering and
Biotechnology, 18(1).
Benjamin P Kleinstiver, Vikram Pattanayak, Michelle S
Prew, Shengdar Q Tsai, Nhu T Nguyen, Zongli Zheng,
J Keith Joung. 2016. High-fidelity CRISPR-Cas9
nucleases with no detectable genome-wide off-taret
effects. Nature, 529(7587), 490-495.
Bon Ham Yip., 2020. Recent Advances in CRISPR/Cas9
Delivery Strategies. Biomolecules, 10(6), 839 page.
Cicera R. Lazzarotto, Nhu T. Nguyen, Xing Tang, Jose
Malagon-Lopez, Jimmy A. Guo, Martin J. Aryee, J.
Keith Joung, Shengdar Q. Tsai. 2018. Defining
CRISPR-Cas9 genome-wide nuclease activities with
CIRCLE-seq. Nat Protocols.
Christine L. Xu, Merry Z. C. Ruan, Vinit B. Mahajan,
Stephen H. Tsang. 2019. Viral delivery systems for
CRISPR. ç, 11(1), 28 page.
Cody S. Lee, Elliot S. Bishop, Ruyi Zhang, Xinyi Yu.,
Evan M. Farina, Shujuan Yan, Chen Zhao, Zongyue
Zeng, Yi Shu, Xingye Wu, Jiayan Lei, Yasha Li,
Wenwen Zhang, ChaoYang, Ke Wu, Ying Wu,
Sherwin Ho, Aravind Athiviraham, MICHAEL J. Lee,
Jennifer Moriatis Wolf, Russell R. Reid and Tong-
Chuan He. 2017. Adenovirus-mediated gene delivery:
Potential applications for gene and cell-based
therapies in the new era of personalized
medicine. Genes & Diseases, 4(2), 43-63.
Elisabeth Kohne. 2011. Hemoglobinopathies: clinical
manifestations, diagnosis, and treatment. Deustsches
Arzteblatt international, 108(31-32), 532-540.
Gael J. Lonergan, David B. Cline, Susan L. Abbondanzo.
2001. Sickle Cell Anemia. RadioGraphics.
Giacomo Frati, Annarita Miccio. 2021. Genome Editing
for beta- Hemoglobinopahties: Advances and
Challenges. Journal of Clinical Medicine.
Jonathan M. Levy, Wei-His Yeh, Nachiket Pendse, Jessie
R. Davis, Erin Hennessey, Rossano Butcher, Luke W.
Koblan, Jason Comander, Qin Liu. and David R. Liu.
2020. Cytosine and adenine base editing of the brain,
liver, retina, heart and skeletal muscle of mice via
adeno-associated viruses. Nature Biomedical
Engineering, 4(1), 97-110.
Killian S. Hanlon, Benjamin P. Kleinstiver, Sara P.
Garcia, Mikolaj P. Zaborowski, Adrienn Volak, Stefan
E. Spirig, Alissa Muller, Alexpander A. Sousa,
Shengdar Q Tsai, Niclas E. Bengtsson, Camilla Lööv,
Martin Ingelsson, Jeffrey S. Chamberlain, David P.
Corey, Martin J. Aryee, J. Keith Joung, Xandra O.
Breakefield, Casey A. Maguire, Bence György. 2019.
High levels of AAV vector integration into CRISPR-
induced DNA breaks. Nature Communications, 10(1).
Lukasz Swiech, Matthias Heidenreich, Abhishek Banerjee,
Naomi Habib, Yinqing Li, John Trombetta, Mriganka
Sur, Feng Zhang. 2014. In vivo interrogation of gene
function in the mammalian brain using CRISPR-Cas9.
Nature Biotechnology, 33(1), 102-106.
Michael C. Milone and Una O’Doherty. 2018. Clinical use
of lentiviral vectors. Leukemia, 32(7), 1529-1541.
Paul S Frenette, George F Atweh. 2007. Sickle Cell
disease: old discoveries, new concepts, and future
promise. The Journal of clinical investigation, 117(4),
850-858.
Renzo Galanello, Raffaella Origa. Beta-thalassemia. 2010.
Orphanet journal of rare diseases, 5, 11.
Suthat Fucharoen, Vip Viprakasit. 2009. Hb H diesase:
clinical courses and disease modifiers. Hematology.
American Society of Hemotalogy Education Program,
26-34.
Shengdar Q Tsai, Zongli Zheng, Nhu T Nguyen, Matthew
Liebers, Ved V Topkar, Vishal Thapar, Nicolas
Wyvekens, Cyd Khayter, A John Iafrate, Long P Le,
Martin J Aryee, J Keith Joung. 2015. GUIDE-seq
enables genome-wide profiling of off-target cleavage
by CRISPR-Cas nucleases. Nat Biotechnol 33, 187-
197.
Shanmuganathan Chandrakasan, Punam Malik. 2014.
Gene therapy for hemoglobinopathies: The state of the
field and the future. Hematol Oncol Clin North Am,
28(2): 199-216.
Stuart H Orkin. 2021. Molecular Medicine: Found in
Translation. Med, 2(2), pp.122-136.
The Protein Man. 2021. CRISPR, ZFNs, TALENs:
Differences Between Bioengineering Technologies.
[online] Info.gbiosciences.com. Available at:
<https://info.gbiosciences.com/blog/crispr-zfns-talens-
differences-between-key-bioengineering-
technologies> [Accessed 1 September 2021].
Vijay G Sankaran, Stuart H Orkin. 2012. The Switch from
Fetal to Adult Hemoglobin. Cold Spring Harbor
Perspectives in Medicine, 3(1), pp. a011643-a011643.
Precision Genome Editing towards the Treatment of Hemoglobinopathies
1075
