
Bioinformatics Helps with Drug Discovery for COVID-19 Treatment
Yicheng Lou
1
, Keyi Shen
2
, Shihhao Fang
3
, Zelu Huang
4
and Yan Lin
5
1
Shanghai East Foreign Language School Affiliated to SISU, Shanghai, 200092, China
2
University of California, Davis, CA 95616, U.S.A.
3
Fudan International School, Shanghai, 200086, China
4
School of Chemical and Biomedical Engineering, Nanyang Technological University, 639798, Singapore
5
Maple Leaf International School-Xi'an, Airport New City, Shaanxi Province, 461000, China
iceylinyan@163.com
Keywords: Drug Discovery, SARS-Cov-2, Spike Protein, Computational Approaches.
Abstract: This paper is a review of the process of drug discovery for COVID-19 treatment based on spike protein. This
work discussed three fundamental approaches: dynamic programming, progressive alignment construction,
and consensus method. Then, the paper used the amino acid sequences (of viral proteins) downloaded from
the UniProt database and applied Clustal Omega (an alignment tool) to conduct multiple alignments on four
spike proteins: SARS-CoV, SARS-CoV-2, MERS-CoV, HCoV-NL63. A brief phylogenetic analysis was also
conducted to support the predicted alignment results. After that, the paper includes a review of applications
of drug discovery based on spike protein alignment—both within the coronaviridae species and with HIV.
1 INTRODUCTION
COVID-19 is a disease caused by the infection of a
novel coronavirus called SARS-CoV-2. Since the
outbreak in December 2019, this devastating
pandemic has widely aroused massive experimental
and analytical investigations into the sophisticated
viral infection pathway and related allopatric treating
methods. Though there existing several approved
vaccines and symptom-oriented treatments for
COVID-19 patients, this might not be enough due to
the high rate of mutation and transmission. Therefore,
there is an increasing demand for potentially more
effective medication to cure the current patients. One
promising method to fulfill this demand is to explore
the existing drugs that may alter the chemical
reactions involving the spike protein of coronavirus.
Spike protein is one of the three major proteins found
in all coronavirus species, which routinely plays an
essential role in penetrating host cells and initiating
the infection process. By applying the multiple
alignment algorithm, similarities can be identified, or
conserved sequences, in the spike proteins within the
coronaviridae family (including SARS, MERS,
SARS-CoV-2). Based on sequence similarity and
information regarding the drugs efficacious against
thoroughly studied coronaviruses such as SARS and
MERS, it is very likely to identify a few multi-
purpose antiviral drugs effective for the treatment of
COVID-19.
2 METHOD
The process of multiple alignment requires input
sequences and an alignment tool. First, from the
UniProt database, amino acid sequences of spike
glycoproteins of four species from the coronaviridae
family were downloaded: SARS-CoV, SARS-CoV-2,
MERS-CoV, and HCoV-NL63. Then, this work used
an alignment tool called Clustal Omega to reveal
similar and conserved segments in their amino acid
sequences.
3 ALIGNMENT ALGORITHMS
AND PHYLOGENY
As the development of computational biology had
boosted in the past few decades, there had been
various methods and algorithms to serve as the
function of multiple sequence alignment. This section
will introduce some fundamental approaches to work
on comparing multiple sequences in the same way.
1104
Lou, Y., Shen, K., Fang, S., Huang, Z. and Lin, Y.
Bioinformatics Helps with Drug Discovery for COVID-19 Treatment.
DOI: 10.5220/0011378100003443
In Proceedings of the 4th International Conference on Biomedical Engineering and Bioinformatics (ICBEB 2022), pages 1104-1111
ISBN: 978-989-758-595-1
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
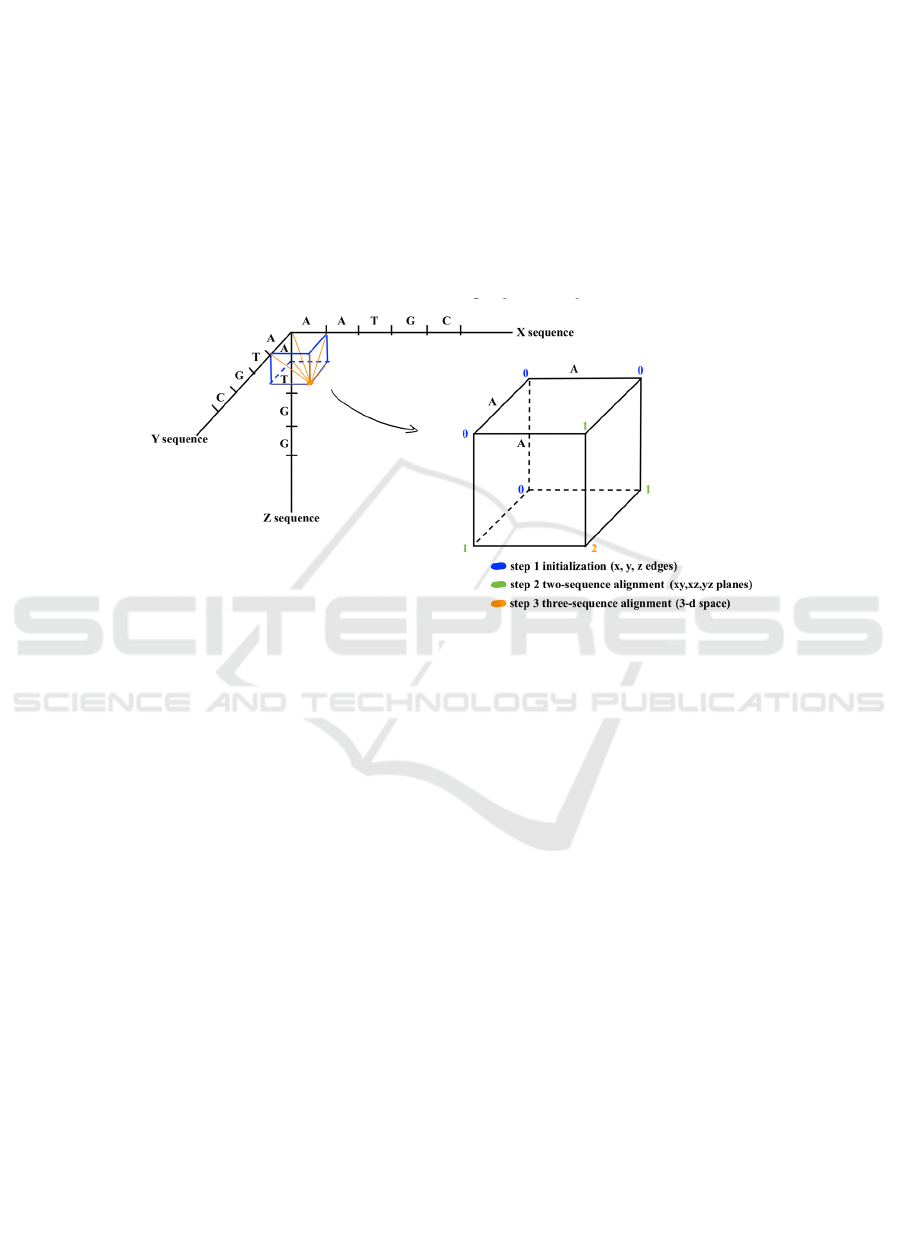
3.1 Dynamic Programming
The disadvantage of using the approach of dynamic
programming is obvious. Even though its logic is
quite easy to understand, and it does work when only
aligning two or three sequences at a time, but its time
and computational complexity make it unsuitable to
compare more than tens or hundreds of sequences
simultaneously. If n sequences with equal length are
compared, the code will have to create an n-
dimensional grid to record the value of the two
nucleotides in every two different sequences with the
same index at the same time. The n-dimensional grid
is also known as the n-Manhattan grid.
(https://www.youtube.com/watch?v=CTPiYiTQcuA
)
Additionally, since node value of node M (a1, a2,
a3, …, an) is determined by the largest value of 2n-1
node values from each of 2n-1 incoming edges, which
each index {an} can choose to minus 1 or remain the
same. This indicates that the program has time
complexity N(2nln), where l stands for the length of
each sequence. Such tremendous complexity makes it
unable to compute and align hundreds of sequences at
the same time. Figure 1 shows how dynamic
programming works.
Figure 1: Working mechanism dynamic programming.
3.2 Progressive Alignment
Construction
The progressive alignment method is a further
improvement of the previous n-Manhattan grid
approach and is the most widely used approach to
multiple sequence alignment (Feng, Doolittle, 1987)
This alignment algorithm is used by Clustal Omega,
an alignment tool used in the upcoming sections. The
core of this method is based on the idea that
combining sequences into profiles before alignment
would not affect the final alignment result. There are
four main steps for this approach. First, every two
sequences in the dataset would be aligned with each
other. Secondly, a distance matrix will be constructed
based on the results for each pair of alignments. Later
on, a phylogenetic tree, or guide tree, will be drawn
on the basis of the distance matrix constructed
previously. Finally, by the guide of the phylogenetic
tree, the program will first process the alignment that
is most similar to each other and combine them into a
profile, which will later participate in the follow-up
combinations and alignments until all the sequences
are combined into one large profile
(https://www.youtube.com/watch?v=CTPiYiTQcuA
)
Progressive alignment methods are efficient enough
for comparing hundred or thousand of sequences at
the same time. The reason is that it only needs to
construct two-dimensional matrices instead of a
massive n-dimensional matrix described in the
previous dynamic programming approach. But still,
its disadvantage is that it is not guaranteed to be
globally optimal, and a small mistake from the
beginning would be inherited and magnified as the
results are combined into profiles and sub-profiles.
Meanwhile, in conditions that each sequence is
dissimilar to another, its performance is not so well.
There are still a lot of space to optimize the scoring
and weighting functions to increase its accuracy.
(Collingridge, Kelly 2012)
3.3 Consensus Method
Despite improving the process of drawing the n-
dimensional Manhattan matrix, scientists also
propose a method to simplify the alignment task:
applying the consensus sequence of each pair of
sequences. Consensus methods attempt to find the
optimal multiple sequence alignments for the given
multiple alignments according to the IUPAC code.
Bioinformatics Helps with Drug Discovery for COVID-19 Treatment
1105

By finding consensus of every two sequences or even
all sequences together, the time and computational
complexity would be greatly decreased. The most
popular consensus-based algorithms are M-COFFEE
and MergeAlign.
3.4 Experimental Alignment of Four S
Protein Sequences
3.4.1 Selection of S Proteins
This work carefully selected S Protein Sequences
from these four viral species for our experimental
alignment, Table 1 facilitates the comparison among
the four species:
1.
SARS-CoV
2.
SARS-CoV-2
3.
MERS-CoV
4.
HCoV-NL63
The primary reason select these four sequences
are selected is that they are similar yet diverse. First
of all, they all belong to the family of coronaviridae.
Second, viruses (1)-(3) belong to the genera of
betacoronavirus while virus (4) belongs to the genera
of alphacoronavirus. However, virus (1), (2), and (4)
has a common receptor called angiotensin-converting
enzyme 2 (ACE-2) whereas virus (3) has a receptor
called Dipeptidyl peptidase-4 (DPP-4) (Totura,
Bavari 2019). In addition, another recent study
suggests that DPP-4 can also act as a receptor of virus
(1) and (2) (Li, 2020). Thus, it is reasonable to make
a hypothesis that the four sequences share some
conserved segments even though they tend to
diversify to a certain degree. Therefore, existing
drugs targeting one of these four proteins (or their
corresponding receptors), may be efficacious against
the rest of them.
Table 1: Comparison of the coronaviruses of our selection.
Species Genera Recepto
r
SARS-CoV-2 betacoronavirus
ACE-2
DPP-4
(
ma
y
act as a co-rece
p
tor
)
SARS-CoV
b
etacoronavirus ACE-2
MERS-CoV
b
etacoronavirus DPP-4
HCoV-NL63 alphacoronavirus ACE-2
3.4.2 Results
Receptor Binding Domain
Figure 2: Receptor binding domain aa alignment result.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1106

According to previous studies, amino acids 323-545
on SARS-CoV-2 spike protein constitute the receptor
binding domain (RBD) (Zhang, 2020); (Pradhan,
2020). Thus, the work focus on this RBD and the
same region in the other three sequences. We
observed that SARS-CoV and SARS-CoV-2 has the
highest level of similarity in their RBD regions, as
shown in Figure 2. Thus, it is reasonable to
hypothesize that SARS-CoV and SARS-CoV-2 can
infect host cells with essentially the same chemical
mechanism. This is confirmed by Zhang et al. by
resolving the molecular structure of S proteins in the
two viral species (Zhang, 2020). In other words, RBD
of SARS-CoV is well preserved in SARS-CoV-2. On
the other hand, unsurprisingly, MERS-CoV and
HCoV-NL63 has significantly lower level of
similarity with SARS-CoV-2 in this region.
Therefore, it is safe to make the prediction that an
existing drug inhibiting the infection of SARS-CoV
is most likely to be efficacious against SARS-CoV-2
as well.
A Brief Discussion on the Phylogeny. Our
prediction can be supported by phylogenetic trees
involving these four viral species. The first step is to
find the distances between species. This work applied
an algorithm called Levenshtein distance
(Levenshtein, 1966). When the input sequences are
limited to their RBD regions, the initial distance
matrix in Table 2 can be obtained. Then, the UPGMA
algorithm is applied for the tree construction (Journal
of Microbiology, 2017). As a result, the phylogenetic
tree obtained is shown in Figure 4.
The edit distance code is shown in Figure 3:
Figure 3: Edit distance code.
Table 2: Initial distance matrix when the input sequences are limited to RBD regions.
SARS-CoV SARS-CoV-2 MERS-CoV HCoV-
NL63
SARS-CoV 0
SARS-CoV-2 94
0
MERS-CoV 184
186 0
HCoV-NL63 170
159 182 0
Bioinformatics Helps with Drug Discovery for COVID-19 Treatment
1107
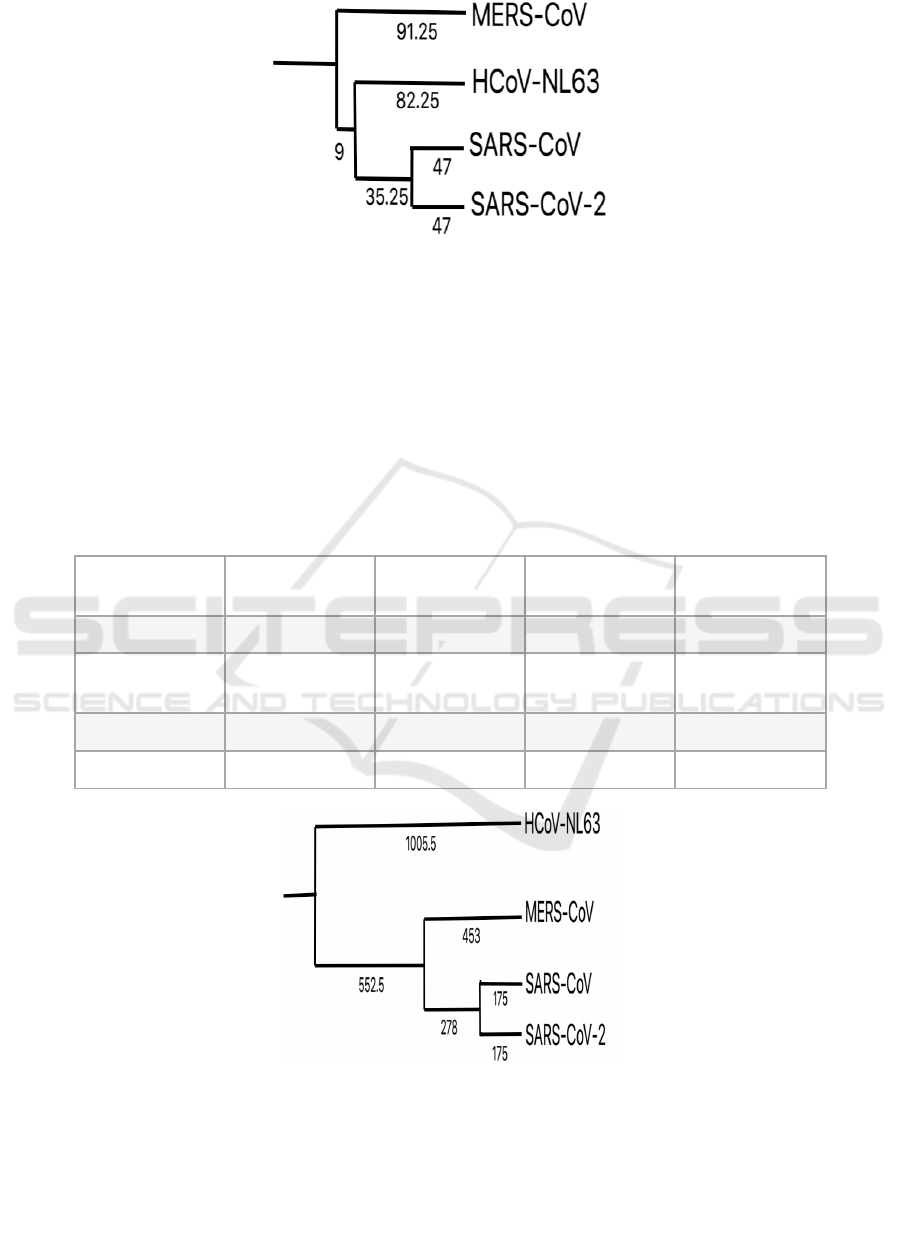
Figure 4: Phylogenetic tree built upon the basis of distance matrix in figure 3.1.
Given that SARS-CoV and SARS-CoV-2 are
betacoronaviruses, it seems surprising that HCoV-
NL63, an alphacoronavirus, is more closely related to
SARS-CoV and SARS-CoV-2 than the MERS-CoV,
a betacoronavirus. This is a result of sample bias
because the input sequences were limited to the RBD
region. Once remove this limit is removed and let the
input sequences be the complete poly-petitde chains,
the distance matrix in Table 3 can be found, and
therefore, construct the phylogenetic tree in Figure 5.
In this less biased tree, HCoV-NL63 is a sister taxon
of all the other viral species. Nevertheless, no matter
which input sequences is used, SARS-CoV is always
the most closely related to SARS-CoV-2 in terms of
similarities in the spike protein. Therefore, it will be
efficient to search for potential drug candidates for
COVID-19 by testing the drugs proved effective to
inhibit the infection of SARS.
Table 3: Distance matrix when the input values are the complete sequences.
SARS-CoV SARS-CoV-2 MERS-CoV HCoV-NL63
SARS-CoV 0
SARS-CoV-2 350 0
MERS-CoV 905 907 0
HCoV-NL63 987 1017 1009 0
Figure 5: Phylogenetic tree based on complete sequences of spike proteins.
Even though this work have already introduced a
potentially efficient method for drug discovery, the
difference between Figure 4 and Figure 5 has not
been discussed. It can be inferred from the difference
in taxa arrangement that the majority of mutations in
the S proteins occur outside their RBD regions. These
mutations are not as important as the ones within the
RBD regions because they do not affect the receptor
binding (or infection) process directly. Thus,
comparing to MERS-CoV, HCoV-NL63 might be
more similar with SARS-CoV-2 in terms of receptor
binding mechanism. Therefore, looking for drug
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1108

candidates among the ones effective for the inhibition
of HCoV-NL63 infection might be the second
efficient method for our drug discovery purpose.
4 DRUG DISCOVERY BASED ON
CONSERVED SPIKE PROTEIN
STRUCTURE
Though different viruses attack different cells and
cause different diseases, there exist some similarities
among different viruses, including similarities in
infection tissue, in transmission vector (Wang, 2020),
and in spike glycoprotein. The latter is the topic that
will be elaborated in this paper. In this part, this work
will identify spike protein structures that are
conserved in multiple coronaviruses. Based on this
information, broad-spectrum antiviral drugs that
might be effective against SARS-CoV -2 can be
found.
4.1 Significance of Spike Protein
For coronavirus, spike protein is a kind of protein on
the surface of the viruses, which is essential for the
virus's combination to receptors. The S protein plays
an essential role in the entry of coronavirus because it
is a class I fusion protein (Wikipedia, 2021).
Moreover, this protein is highly related to antibody
production (Hughes, 2011), making it a crucial focus
on researches related to COVID-19 diagnosis.
What is focused in this paper is how spike protein
of coronavirus can be applied to potential drug
discovery. Efficacy of drugs for certain viral spike
protein may be extended to viruses with similar spike
protein structure. Thus, drug that's proved efficacious
against one or a few species in the coronaviridae
group (e.g. SARS CoV or MERS-CoV) can be
identified, it is very likely to find that the drug is also
effective for the treatment of COVID-19. Moreover,
such approaches of drug discovery is efficient and
less time-costing compared to traditional drug
discovery approaches (Kumar, S, 2020). Therefore,
drug discovery based on spike protein has great
potential in finding drugs during the COVID-19
pandemic.
4.2 Current Applications in Drug
Discovery
After the viral genome of SARS-CoV-2 is released on
10 January 2020, there has been efforts in drug
discovery based on spike protein and its receptors.
Scientists have analyzed chemical characteristics
of the spike protein from the perspective of molecular
weight, number of amino acids, electricity charge,
etc. (Seresh Kumar) (Ibrahim, Ibrahim M, 2020).
4.2.1 Receptor Inhibition
In order to prevent the spike protein of SARS-CoV-2
from binding to ACE-2 receptor. Drugs can either
inhibit the spike protein or inhibit the receptor. As it
is stated in Section 4 of Part III, if a drug can inhibit
the binding between spike protein of SARS-CoV and
ACE-2 receptor, it is very likely to be efficacious
against SARS-CoV-2 by the same mechanism. For
instance, as early as 2005, a group of scientists
discovered in vivo that hydroxychloroquine (HCQ) is
able to inhibit the binding between the spike protein
of SARS-CoV and ACE-2 by interrupting the
glycocylation of ACE-2 in the Golgi apparatus, which
changes the structure of ACE-2 (Vincent, M, 2005).
Inspired by this study, this work reviewed a major
clinical trial on therapeutic use of HCQ in COVID-19
infection (Million, 2020). This study indicates that
over 95% of the patients treated with HCQ were cured
on the tenth day of treatment, and only 2.3% of them
reported mild adverse effects (Million, 2020). Thus,
HCQ seems to be a promising drug for the COVID-
19 treatment. This discovery implies that our drug
discovery strategy is potentially effective.
4.2.2 Spike Protein Inhibition
Through predicting the genome encoding for spike
protein and comparing the SARS-CoV-2 spike
protein with number of database, the research stated
that the spike protein of SARS-CoV-2 has high
similarity with SARS, and that inhibitors of 3C-like
protease(3CLpro) is a potentially effective anti-viral
drug (Kliger, 2003).
Ibrahim M et al. used combined molecular
docking studies and made the prediction that the spike
protein of SARS-CoV-2 can bind to GRP78(Glucose
Regulated Protein 78), which is a host receptor for
SARS (Sokal, Mechener, 1958). Thus, antiviral drugs
for GRP78, which are effective for treating SARS
infections, might be a potential candidate for COVID-
19 treatment (Feng, Doolittle, 1987).
Outside the species of coronaviridae, scientists are
discovering similarities between spike protein of
SARS-CoV-2 and spike protein of another virus--the
HIV. Kliger, Y and Levanon, E.Y applied heptad
repeat analysis and transmembrane domain
prediction to find out similarity between fusion
proteins of SARS-CoV-2 and HIV. However,
sequence comparison algorithms showed little
Bioinformatics Helps with Drug Discovery for COVID-19 Treatment
1109

similarity between the two fusion proteins. The
similarity only exists in the aspect of mechanism, so
that the two fusion proteins are analogous to each
other (Collingridge, Kelly, 2012). This suggests that
drugs effective for repressing HIV fusion, such as
C34, may also repress the fusion of SARS-CoV-2
with host cells. Moreover, this research broadens our
consideration, providing another perspective--
analogous proteins.
4.2.3 Discussion
One things all these applications mentioned is that
biological studies and lab experiments are needed.
Some of the researches have been proved by
experimental data while some others haven't. It is
important to keep in mind that though bioinformatics
approaches are effective, they should always be
proved by lab experiments.
5 CONCLUSION
With the help of bioinformatics, the drug discovery
process based on spike protein multiple alignment is
becoming an effective solution facing the urgent need
for drugs during the COVID-19 pandemic. Multiple
alignment procedure can be verified by applying
several computational approaches or by using
phylogenetic analysis. Such process of drug
discovery reduces both time and costs. Moreover,
besides spike protein alignment within the
coronaviridae species, comparing spike protein of
coronavirus with other viral species, such as HIV,
provides us with new insights. For drug design based
on spike protein, following laboratory experiments
are essential. This work can be a source for future
investigations, including lab verifications and clinical
studies.
REFERENCES
Collingridge PW, Kelly S (2012). "MergeAlign: improving
multiple sequence alignment performance by dynamic
reconstruction of consensus multiple sequence
alignments". BMC Bioinformatics. 13 (117): 117.
doi:10.1186/1471-2105-13-117. PMC 3413523. PMID
22646090.
Collingridge PW, Kelly S (2012). "MergeAlign: improving
multiple sequence alignment performance by dynamic
reconstruction of consensus multiple sequence
alignments". BMC Bioinformatics. 13 (117): 117.
doi:10.1186/1471-2105-13-117. PMC 3413523. PMID
22646090.
Feng DF, Doolittle RF (1987). "Progressive sequence
alignment as a prerequisitetto correct phylogenetic
trees". J Mol Evol. 25 (4): 351–360.
Bibcode:1987JMolE..25..351F.
doi:10.1007/BF02603120. PMID 3118049. S2CID
6345432.
Feng DF, Doolittle RF (1987). "Progressive sequence
alignment as a prerequisitetto correct phylogenetic
trees". J Mol Evol. 25 (4): 351–360.
Bibcode:1987JMolE..25..351F.
doi:10.1007/BF02603120. PMID 3118049. S2CID
6345432.
https://www.youtube.com/watch?v=CTPiYiTQcuA
https://www.youtube.com/watch?v=CTPiYiTQcuA
Hughes, J.P. et al. Principles of early drug discovery.,
British Journal of Pharmacology. (2011)
Ibrahim, Ibrahim M et al. “COVID-19 spike-host cell
receptor GRP78 binding site prediction.” The Journal
of infection vol. 80,5 (2020): 554-562.
doi:10.1016/j.jinf.2020.02.026
Journal of Microbiology (2017) Vol. 55, No. 2, pp. 81–89
Kumar, S. Drug and Vaccine Design against Novel
Coronavirus (2019-nCoV) Spike Protein through
Computational Approach. Preprints 2020, 2020020071
(DOI: 10.20944/preprints202002. 0071.v1).
Kliger, Y., Levanon, E.Y. Cloaked similarity between HIV-
1 and SARS-CoV suggests an anti-SARS strategy.
BMC Microbiol 3, 20 (2003).
https://doi.org/10.1186/1471-2180-3-20
Li, Y. et al. (2020) "The MERS-CoV Receptor DPP4 as a
Candidate Binding Target of the SARS-CoV-2 Spike."
iScience Vol. 23, Issue 6
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7219414/
Levenshtein, V. (1966) "Binary codes capable of correcting
deletions, insertions, and reversals." Soviet Physics
Doklady (in Russian). Vol. 10, Issue 8, pp. 707–710
http://www.mathnet.ru/links/984d857b9221e734b627f0a9
45fd3d7e/dan31411.pdf
Million, M. et al. (2020) “Early treatment of COVID-19
patients with hydroxy- chloroquine and azithromycin:
A retrospective analysis of 1061 cases in Marseille,
France”. Travel Medicine and Infectious Disease.
Volume 35. Retrieved from:
https://www.sciencedirect.com/science/article/pii/S147789
3920302179?via%3Dihub
Million, M. et al. (2020) “Early treatment of COVID-19
patients with hydroxy- chloroquine and azithromycin:
A retrospective analysis of 1061 cases in Marseille,
France”. Travel Medicine and Infectious Disease.
Volume 35. Retrieved from:
https://www.sciencedirect.com/science/article/pii/S147789
3920302179?via%3Dihub
Pradhan, P. et al. (2020) "Uncanny similarity of unique
inserts in the 2019-nCoV spike protein to HIV-1 gp120
and Gag." BioRxiv
https://www.biorxiv.org/content/10.1101/2020.01.30.9278
71v1
Sokal, R. and Mechener, C. (1958) "A statistical method for
evaluating systematic relationships." University of
Kansas Science Bulletin. Vol. 38, Issue 2, pp. 1409-
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1110

1438
https://archive.org/details/cbarchive_33927_astatisticalmet
hodforevaluatin1902/page/n27/mode/2up
Totura, A. and Bavari S. (2019) "Broad-spectrum
coronavirus antiviral drug discovery." Expert Opinion
on Drug Discovery Vol. 14, Issue 4, pp.397-412
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7103675/
Vincent, M. et al. (2005) “Chloroquine is a potent inhibitor
of SARS coronavirus infection and spread” Virology
Journal Volume 2. Issue 29.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1232869/
Wang, Yuhang et al. “Coronaviruses: An Updated
Overview of Their Replication and Pathogenesis.”
Methods in molecular biology (Clifton, N.J.) vol. 2203
(2020): 1-29. doi:10.1007/978-1-0716-0900-2_1
Wikipedia contributors. "Spike protein." Wikipedia, The
Free Encyclopedia. Wikipedia, The Free Encyclopedia,
6 Sep. 2021. Web. 6 Sep. 2021.
Zhang, C. et al. (2020) "Protein Structure and Sequence
Reanalysis of 2019-nCoV Genome Refutes Snakes as
Its Intermediate Host and the Unique Similarity
between Its Spike Protein Insertions and HIV-1."
Journal of Proteome. Vol. 19, Issue 4, pp. 1351-1360
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7099673/
Zhang, C. et al. (2020) "Protein Structure and Sequence
Reanalysis of 2019-nCoV Genome Refutes Snakes as
Its Intermediate Host and the Unique Similarity
between Its Spike Protein Insertions and HIV-1."
Journal of Proteome. Vol. 19, Issue 4, pp. 1351-1360
Bioinformatics Helps with Drug Discovery for COVID-19 Treatment
1111
