
The Phylogenetic Analysis of 5 SARS-CoV-2 Proteins’ Sequences in
Relation to Time and Geographical Location
Sophia Ying Li
1,†
, Xiayan Li
2,*
, Jiahe Gao
3
, Kaiyang Pang
4
and Deyuan Xu
5
1
Thomas Jefferson High School for Science and Technology, 6560 Braddock Rd Alexandria, VA 22312, U.S.A.
2
School of Computer science and Engineering, Baylor University, 1311 S 5th St, Waco, TX 76706, U.S.A.
3
Qingdao No.2 High School, Qingdao, Shandong, 266061, China
4
Revelle College, University of California San Diego, 9500 Gilman Drive, La Jolla, 92093, U.S.A.
5
College of Bioinformatics Science and Technology, Harbin Medical University, Harbin, Heilongjiang, 150076, China
Keywords: Bioinformatics, Protein Sequence, SARS-Cov-2, Geographical Location, Phylogenetic Analysis, Evolution.
Abstract: After 18 months since the start of the COVID-19 epidemic, the virus continues to plague the world. Learning
more about how SARS-CoV-2 proteins mutate and the relation to spatial location is critical in helping us
predict the spread of variants. We built an MSA and UPGMA phylogenetic tree for each of the 5 crucial ORF
sequences (S, M, E, N, and ORF1ab protein), which were collected by the latest update date for each region,
and based on the results, we are not able to conclude that SARS-CoV-2 protein sequences from different
countries co-evolved with other SARS-CoV-2 protein variants in proximity. However, some highly mutated
regions within those sequences may suggest some evolutionary pattern during this continuing pandemic.
1 INTRODUCTION
1.1 Spread of Coronavirus
The outbreak of severe acute respiratory syndrome
coronavirus (SARS-CoV-2) across the globe has had
devastating impacts on various countries. In
December 2019, the first COVID-19 infection, the
disease caused by SARS-CoV2, was reported in
Wuhan, China (Wang, Horby, Hayden, Gao 2020).
The number of infections reports then increased
rapidly around the world. In March 2020, COVID-19
was declared as a global pandemic by The World
Health Organization (WHO) (“WHO Director-
General’s opening remarks at the media briefing on
COVID-19 - 11 March 2020)
As of September 2021,
the CDC has reported almost 40 million cases and
over 600 thousand deaths in the United States of
America (CDC 2021). There has been a total of
around 218.9 million cases and 4.5 million deaths
(WHO Coronavirus (COVID-19) Dashboard).
Although the epidemic is currently under control,
COVID-19 is still spreading in some countries and
areas, threatening global health systems and health
security. For the foreseeable future, novel coronavirus
outbreaks will continue for several years, and national
and regional prevention measures will continue.
Novel coronavirus, a part of the Coronaviridae
family, causes severe respiratory infections in
mammals. According to the World Health
Organization, based on the accumulated
observations, the most common symptoms of
COVID-19 are fever, dry cough, and fatigue. Less
common symptoms include loss of taste or smell,
nasal congestion, conjunctivitis (also known as red
eyes), sore throat, headache, muscle or joint pain, and
nausea (Coronavirus disease (COVID-19).”). There
have also been many asymptomatic cases, as seen
through a study of healthcare workers by Wilder-
Smith et al (A. Wilder-Smith, M. D. Teleman, B. H.
Heng, A. Earnest, A. E. Ling, and Y. S. Leo 2005).
1.2 Geographical Location and
Mutations of SARS-Cov2
One study by Fan et al. predicted the outbreak to
come from bats and China because of the data from
past SARS-related coronavirus outbreaks. The study
predicts hotspots for the emergence of the virus using
3 factors: recombination from rich gene pools, the
distance between bats and humans, and virus
transmissibility. The researchers found the spread of
many diverse and closely related CoVs between bats
1164
Li, S., Li, X., Gao, J., Pang, K. and Xu, D.
The Phylogenetic Analysis of 5 SARS-CoV-2 Proteins’ Sequences in Relation to Time and Geographical Location.
DOI: 10.5220/0011381500003443
In Proceedings of the 4th International Conference on Biomedical Engineering and Bioinformatics (ICBEB 2022), pages 1164-1173
ISBN: 978-989-758-595-1
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

of various provinces in China (Fan, Zhao, Shi, Zhou
2019). Their result indicated a positive relationship
between short spatial distances and bat coronavirus
mutation rate.
1.3 Coronavirus Proteins
Novel coronavirus is a single-stranded RNA virus
that consists of 4 structural proteins: surface or spike
glycoprotein (S), membrane glycoprotein (M),
envelope protein (E), and nucleocapsid
phosphoprotein (N). In the SARS-CoV2 genome,
there are 10 operating reading frames (ORF), and the
first ORF (ORF1ab) encodes for 1-16 non-structural
proteins and phosphoprotein a and b, representing the
biggest gene in the coronavirus’s genome (Satarker,
Nampoothiri 2020). The unique sequences which
ORF1ab contains have been recognized as potential
early detection targets for novel Coronavirus (Corman
et al 2020, Jung et al 2020, Wang et al 2020). The rest
code for the structural and accessory proteins. The M
protein is present in high amounts and helps mediate
the inflammatory response in hosts. The E protein is
a tiny integral membrane protein that enhances viral
pathogenicity and aids in virion assembly by
producing viroporins. The N protein enhances viral
entry and plays a critical part in virus transcription
and assembly (McBride, M. van Zyl, Fielding 2014).
The S protein is also known as surface glycoprotein or
spike protein. It plays an important role in
conformational rearrangement to membrane fusion,
which creates pores on the host transmembrane that
viral genomes can be passed through during the viral
transmission. Specifically, the peptide 353- KGDFR-
357 (H. sapiens ACE2 residue numbering), located on
the surface of the ACE2 molecule, participates in the
binding of the SARS-CoV-2 receptor-binding
domain (RBD) (Huang, Yang, Xu, Xu, Liu 2020).
The ACE2 receptor is expressed in lung, intestine,
kidney, and epithelial alveolar type II cells.
13
Therefore, the study of mutation patterns within S
protein may be crucial in understanding virus
reproductive adaptability and evolutionary survival in
fast. These proteins are the building blocks of the
virus, and understanding their sequence is crucial in
understanding their function. However, due to the
high mutation capacity of SARS-CoV-2 and its
increasing adaptability to the environment, more
research is still needed to eliminate the effects of the
virus and restore the normal functioning of society. In
particular, the Delta variant has been spreading faster
than other variants due to its reduced sensitivity to
antibodies that target the S proteins (Planas et al
2021). This study focuses on understanding the
evolutionary pattern among the ORF1ab, S, M, E, and
N of SARS-Cov2 from different countries. By
comparing multiple sequence alignment (MSA) and
analyzing phylogenetic trees, this study aims to
discover the correlation between mutation rates and
geographical location. Specifically, this includes the
results suggesting whether specific protein sequences
are conserved or highly mutated depending on the
region of origin.
2 RESULTS
2.1 Minimal Relationship between
Mutation of Different Proteins and
Geographical Location
Based on the 5 protein UPGMA phylogenetic trees
(see Figure 1-2), there is minimal correlation between
specific protein mutation rates and geographical
location. Moreover, there seems to be no relationship
between S, M, E, Orf1ab, and N. Each tree has a
significantly different structure based on which
region each protein sequence came from. This means
these proteins do not evolve with one another.
The Phylogenetic Analysis of 5 SARS-CoV-2 Proteins’ Sequences in Relation to Time and Geographical Location
1165

(a) UPGMA phylogenetic tree of envelope protein (E)
(b) UPGMA phylogenetic tree of membrane glycoprotein (M)
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1166

(c) UPGMA phylogenetic tree of ORF1ab
(d) UPGMA phylogenetic tree of surface Nucleocapsid protein (N)
Figure 1: UPGMA phylogenetic tree of relatively conserved SARS-CoV-2 proteins’ sequences from various countries.
The Phylogenetic Analysis of 5 SARS-CoV-2 Proteins’ Sequences in Relation to Time and Geographical Location
1167

2.2 MSAs Indicate There to Be No
Specific Mutation Point
There are no major similarities between where
mutations of amino acids occur in any MSAs of each
protein [see Figure 3-6]. The difference between each
region’s amino acid sequences is relatively random
indicating there are no mutation points within any of
the 5 proteins. However, this does not mean that there
are no differences between the sequences. Each
protein has variations between sequences from
different countries, and S protein evolves and mutates
most of the 5 proteins analyzed.
2.3 M, ORF1ab, N, and E Protein Are
Highly Conserved between
SARS-Cov-2 of Different Countries
The phylogenetic tree of M protein has few branches,
and there are only a few differences between the M
protein sequences of the 89 countries. The one
major variation is at position 82 aa of 222 aa where
16 sequences have threonine, 70 have isoleucine, and
2 have serine. Serine and threonine are polar amino
acids, while isoleucine is nonpolar. This may affect
the structure of the protein. 3D structure prediction is
needed to understand the impact of this variation.
Unlike the other countries’ sequences, the sequence
from Iraq and Cameroon has a long section of
’undetermined or atypical amino acids’ near the
beginning and the end respectively. Overall, the
phylogenetic tree and MSA suggest that the M protein
sequence is highly conserved. For ORF1ab, there is
one highly mutated region observed from the MSA at
3667 aa - 3691 aa, and there is a gap introduced in
Lebanon, Philippines, Venezuela, Ghana, Taiwan,
Djibouti, Cambodia, Poland, Dominican Republic,
Tunisia, Saudi Arabia, South Africa, France, Togo.
However, this region is not fully studied, so it is hard
to draw any conclusion on the contribution of this
region to the evolutionary pattern. Sequences of N
protein were analyzed, and the mutation rate is
relatively high at 503 aa. In most protein sequences,
the amino acid at this position would be asparagine.
However, there are 15 sequences where the amino
acid of this position is tyrosine. By extracting the
table corresponding to the countries of those 15
sequences, it turns out that 13 of those sequences
come from mid-latitude regions if not low-latitude
regions. These countries are mainly under the
influence of tropical climate or subtropical climates.
According to the MSA analysis, sequences of E
protein in different regions are comparatively similar.
Moreover, because there are also only a few branches
in the phylogenetic tree, E protein is likely to have a
highly conserved sequence between different
countries. The most noticeable differences are ones in
a few tropical countries: Somalia, Saudi Arabia, and
Kenya. They follow the highly conserved consistency
and mutate only in particular regions. The biggest
difference is a mismatch at 71 aa, in which 5 countries
have leucine and the rest have proline.
2.4 S Protein Is Highly Mutated
between SARS-Cov-2 of Different
Countries
Figure 2: UPGMA phylogenetic tree of highly mutated
SARS-CoV-2 of surface glycoprotein(S).
There are numerous gaps and mismatches detected
from the MSA outcome of S protein: gaps are detected
from 57 aa - 263 aa and 682 aa - 686 aa (this gap is
mainly caused by the 4 aa insertions from the Russia
sequence), and few mismatches are detected at the
regions close to the gaps. This may suggest these
regions undergo positive evolution. Within the
UPGMA Phylogenetic tree graph, the distance unit,
which is 0.001, for this tree is relatively larger than
the M, N, E, and ORF1ab protein. Surprisingly, the
sequence from Russia has a long-stretched branch at
the beginning of the root tree, which may be caused
by 4 aa insertions from 682 aa - 686 aa, and the other
region sequences which have introduced gaps, also are
at the position close to the root. There are also no
obvious spatial or time relations captured within the
protein S phylogenetic tree.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1168
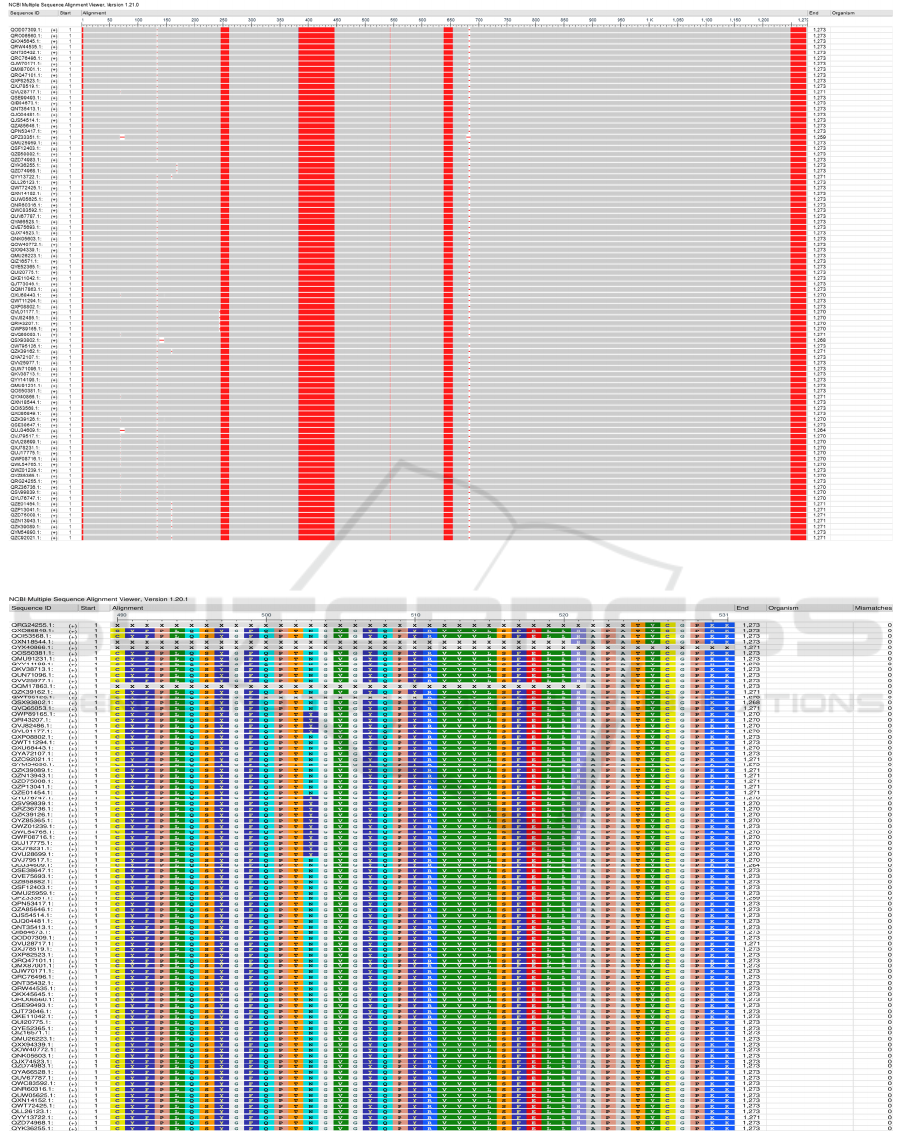
Figure 3: Protein S frequency-based difference MSA.
Figure 4: MSA snapshot of protein N.
The Phylogenetic Analysis of 5 SARS-CoV-2 Proteins’ Sequences in Relation to Time and Geographical Location
1169
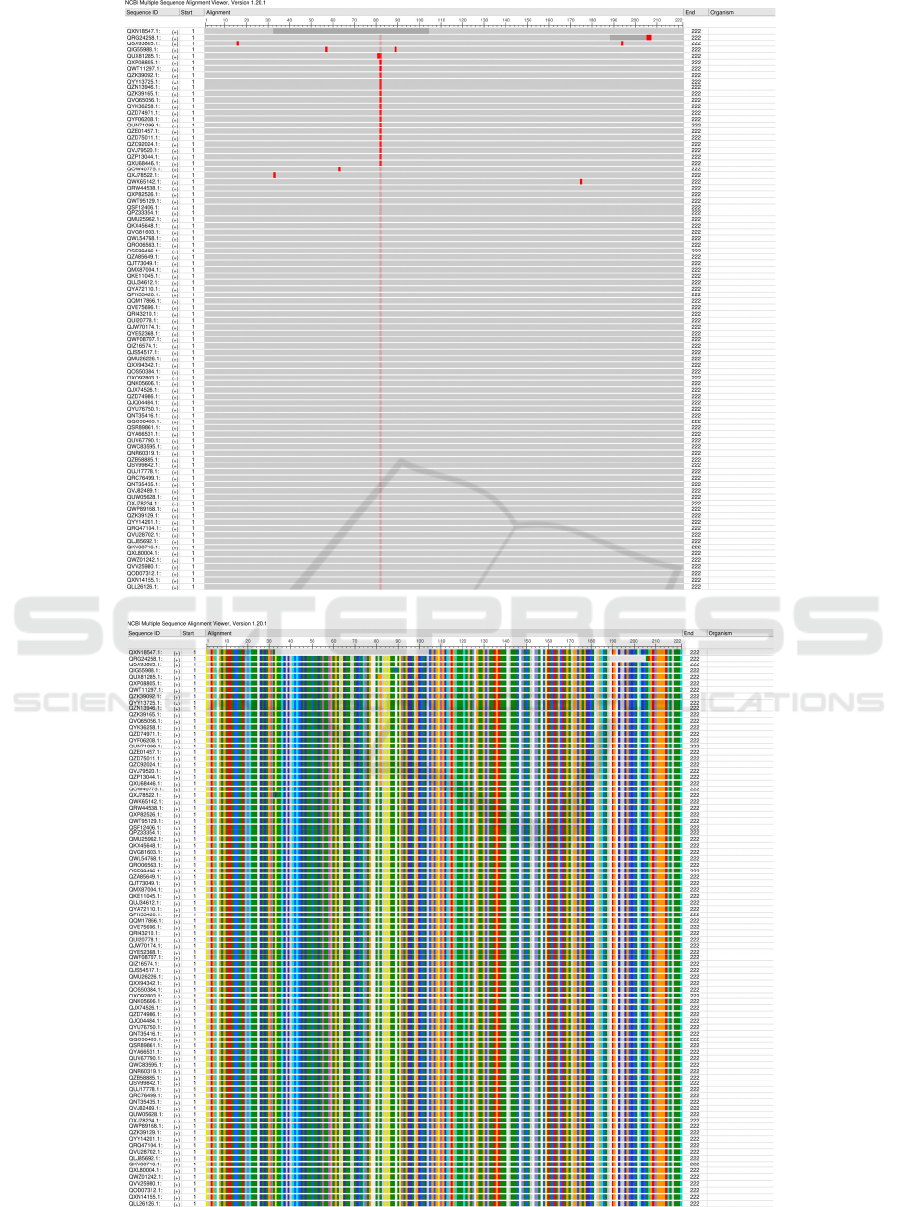
Figure 5: Protein M frequency-based difference MSA.
Figure 6: Protein M rasmol amino acid coloring.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1170

3 DISCUSSION
3.1 Geography and Time Have an
Insignificant Role in SARS-Cov-2
Evolution Pattern
In the UPGMA phylogenetic tree of the 5 proteins, it
is hard to conclude that location and time play a role
in the SARS-CoV-2 evolutionary path. For instance,
the latest sequences for countries in North America on
the S protein phylogenetic tree is scattered: USA is on
branch 17, but Canada is on branch 0. Moreover, the
protein sequence QNT 35432.1, Puerto Rico,
2020/09/18 updated in 2020 is not close to the root,
which may be due to global traveling. Traveling
makes the contact of individuals from non-
neighboring regions easier, so the study of sequences
from a region with less traveling may help eliminate
this factor. Moreover, this introduction of a new
variant may be the reason that unrelated protein
sequences are similar. Further studies are needed to
understand the mechanisms related to the mutation
rates of SARS-CoV-2 proteins. A similar study on
SARS-CoV SNVs (Single nucleotides variations)
also suggests that SNVs frequency across different
regions may be different by geography and time
(Chen, Altschuler, Zhan, Chan, Deverman 2021).
Therefore, there are minimal effects from geography
and time on the small-scale (nucleotide) evolutionary
pattern, and this result may be similar to the minimal
effects of geography and time on a larger scale
(sequences).
3.2 Difference between Highly Mutated
and Conserved Proteins
The phylogenetic tree of the ORF1ab protein has a
smaller scale compared to the phylogenetic tree of
other proteins. Because this protein consists of 2/3 of
the RNA and encodes for the nonstructural proteins,
the results strongly suggest the sequence of this
protein is highly conserved between SARS-CoV-2 of
different countries. Considering the function of N and
E protein have with the virus assembly, the process is
highly vulnerable to the temperature of the
environment. In other words, the difference occurring
at 503 aa of N protein and 71 aa of E protein may be
the adaptivity of the proteins to the climate, further
altering the virus’ survival and spreading under
different climates. More research on the temperature
sensitivity of N protein is needed to understand this
difference. The highly conserved nature of M
proteins is likely related to its function in helping the
virus survive in host cells as it inhibits NFB (Nuclear
Factor Kappa B), which needs to be activated to
produce an immune response to pathogens. This
process is specific because of the direct interaction
between M protein and Iκκβ (I Kappa B Kinase)
(Fang et al 2007). Noticeably, Protein S MSA
outcome reveals two highly mutated regions occurred
from 57 aa - 263 aa and 682 aa - 686 aa in sequence,
and those highly mutated regions are found in the N-
terminal domain (NTD) of the S1 subunit and S1/S2
cleavage regions. The second highly mutated regions
are 6 - 10 aa upstream of the cleavage site, which this
site should be cleaved during virus egress, and highly
mutation pattern captured within this site may suggest
that the variability of this region provide a gain-of-
function to the SARS-CoV-2 for efficient spreading in
the human population compared to other lineage b
beta coronaviruses (Coutard 2020). Since the
comparison made is between countries, it may also
suggest that this virus is highly adaptable within a
changing environment. Additionally, evidence shows
that NTD of the S1 subunit is involved in promoting
cytokine release in immune cells, and this appearance
of cytokines can lead to respiratory failure and a fatal
outcome (Chan et al 2021). Thus, the frequent
mutation within NTD-S1 subunit regions may give
rise to varying severity of immune response across the
different mutant strains and adaptive attack pathways
towards different races of peoples’ immune systems.
In general, the mutation pattern of SARS-CoV-2
Protein S may assist the rapid spreading rate and acute
immune response during the evolution process.
3.3 Future Direction
Combining the study of human immune response
variants towards SARS-CoV-2 may be conse
quential
for understanding how the mutations get selected in the
co-evolution with the host immune
system
mechanisms, and the patient’s record from the
GWAS catalog can be a valuable resource for
analysis. In addition, understanding how these
mutations affect each of the protein structures will be
crucial in finding further therapeutic options and
improving vaccine booster shots.
4 METHODS
4.1 Protein Sequences Collection
The latest sequences of ORF1ab polyprotein, spike
glycoprotein, envelope protein, membrane
glycoprotein, nucleocapsid phosphoprotein were
The Phylogenetic Analysis of 5 SARS-CoV-2 Proteins’ Sequences in Relation to Time and Geographical Location
1171

collected for each region (“INSDC Country List.) if
the data was present. In this study, these sequences
were collected by a script, which is available at
github repository. Entrez-direct (Kans 2021) an Unix
command-line tool that provides access to the NCBI
interconnected database, was used to collect the
protein ID, regions name and update dates, and
download sequences in FASTA files format. There
were 88 sequences collected for ORF1ab, 91
sequences collected for protein S, 89 sequences
collected for Protein E, 89 sequences collected for
Protein M, and 89 sequences collected for Protein N.
The header for each sequence in the FASTA file was
substituted into the format of “protein id:region name
updateDate” to produce straightforward vi-
sualization for further analysis, and “fixed
sequences.fasta” file stores the fixed header and
amino acids sequence. All sequences for five proteins
were collected on 09/04/2021, and the MSA and
Phylogenetics Tree analyses were made on the same
day.
4.2 MSA Analysis
The five sequences FASTA files were separately
aligned by MUSCLE, a tool for creating multi- ple
alignments of protein sequences in high biological
accuracy and time efficiency (Edgar 2004). No
special parameters were set except the input file and
output file because the alignments were relatively
short and there were few alignments. After running
MUSCLE on a five sequence FASTA file, it
produced the alignments files which insert gaps to
achieve the maximum sum-of-pairs (SP) score. The 5
alignment files were the input of the MEGA X tool to
make the UPGMA phylogenetic trees. Because it was
hard to capture the mismatch and gaps in 5 alignment
files, NCBI Multiple Se- quence Alignment Viewer
(NCBI Multiple Sequence Alignment Viewer) a
graphical display for multiple alignments of
nucleotide and protein sequences, is used to produce a
better graphic visualization of MSA analysis results.
The partial or whole scope of MSA outcome was
captured to show the significant regions.
4.3 UPGMA Phylogenetics Tree
Analysis
Finally, the UPGMA phylogenetic trees were created
by MEGA X (Kumar, Stecher, Li, Knyaz, Tamura
2018) a software that implements tools for
phylogenomic analysis. ‘Bootstrap method’ -> ‘Test
of Phylogeny’, ‘500’ -> ‘No. of Bootstrap
Replication’, ‘Amino Acid’ -> ‘Substitutions Type’,
Poisson model’ -> ‘Model/Method’, ‘Uniform Rates’
-> ‘Rates among sites’, ‘same (Homogeneous)’ ->
‘Pattern among Lineages’, and ‘Pairwise deletion’-
>’Gaps/Missing Data Treatment’ were set for the
progress, and rooted trees were kept in a circle
format.
5 CONCLUSION
By analyzing the UPGMA phylogenetic tree and
MSAs of each protein, the results show that
geographical location has an insignificant impact on
SARS-CoV-2 protein mutations and relationships. A
few potential highly mutated regions among the
sequence of each region may suggest the dynamic
adaptivity to diverse environments and alternative
invading strategies to human immune response.
Because geographical location and time do not have a
direct relationship to SARS-CoV-2 protein
mutations, there may be a more complex underlying
factor to explain the relationships between the
protein sequences of various SARS-CoV-2 which can
be studied in the future. Carriers such as different
animals and humans may be one of the many factors
that contribute to this similarity between protein
sequences. Variability in the human immune system
may also be a fac- tor that causes indirect relations
between temporal, geographical location, and SARS-
CoV-2-point mutation patterns. Therefore, more
studies about how the human immune system evolved
during the pandemic should be analyzed. This could
be done by researching patients’ variants record from
the GWAS catalog to prove the correlation with the
variability of the human immune system.
ACKNOWLEDGMENTS
Author superscripts are ordered by number for
contribution and letter for alphabetical order by
authors’ name. Sophia Ying Li and Xiayan Li are the
first co-authors (marked by plus sign), and Jiahe Gao,
Kaiyang Pang, and Deyuan Xu are the second co-
authors.
LIST OF FIGURES
1
UPGMA phylogenetic tree of relatively
2
UPGMA phylogenetic tree of highly mutated
SARS-CoV-2 of surface glycoprotein (S).
3
MSA Overview of protein S
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1172

4
MSA snapshot of protein N
5
Protein M frequency-based difference MSA
6
Protein M rasmol amino acid coloring
REFERENCES
“Coronavirus disease (COVID-19).”
“INSDC Country List.”
“NCBI Multiple Sequence Alignment Viewer 1.21.0.”
“WHO Coronavirus (COVID-19) Dashboard.”
“WHO Director-General’s opening remarks at the media
briefing on COVID-19 - 11 March 2020.”
A. T. Chen, K. Altschuler, S. H. Zhan, Y. A. Chan, and B.
E. Deverman, “COVID-19 CG enables SARS-CoV-2
mutation and lineage tracking by locations and dates of
interest,” eLife, vol. 10, p. e63409, Feb. 2021.
A. Wilder-Smith, M. D. Teleman, B. H. Heng, A. Earnest,
A. E. Ling, and Y. S. Leo, “Asymp- tomatic SARS
Coronavirus Infection among Healthcare Workers,
Singapore,” Emerging In- fectious Diseases, vol. 11,
pp. 1142–1145, July 2005.
B. Coutard, C. Valle, X. de Lamballerie, B. Canard, N.
Seidah, and E. Decroly, “The spike glycoprotein of the
new coronavirus 2019-nCoV contains a furin-like
cleavage site absent in CoV of the same clade,”
Antiviral Research, vol. 176, p. 104742, Apr. 2020.
C. Wang, P. W. Horby, F. G. Hayden, and G. F. Gao, “A
novel coronavirus outbreak of global health concern,”
The Lancet, vol. 395, pp. 470–473, Feb. 2020.
CDC, “COVID Data Tracker Weekly Review,” Nov. 2021.
D. Planas, D. Veyer, A. Baidaliuk, I. Staropoli, F. Guivel-
Benhassine, M. M. Rajah, C. Plan- chais, F. Porrot, N.
Robillard, J. Puech, M. Prot, F. Gallais, P. Gantner, A.
Velay, J. Le Guen,
H. Wang, X. Li, T. Li, S. Zhang, L. Wang, X. Wu, and J.
Liu, “The genetic sequence, origin, and diagnosis of
SARS-CoV-2,” European Journal of Clinical
Microbiology & Infectious Diseases: Official
Publication of the European Society of Clinical
Microbiology, vol. 39, pp. 1629–1635, Sept. 2020.
J. Kans, Entrez Direct: E-utilities on the Unix Command
Line. National Center for Biotech- nology Information
(US), Nov. 2021. Publication Title: Entrez
Programming Utilities Help [Internet].
M. Chan, S. Vijay, J. McNevin, M. J. McElrath, E. C.
Holland, and T. S. Gu- jral, “Machine learning
identifies molecular regulators and therapeutics for
targeting SARS[U+2010] CoV2[U+2010] induced
cytokine release,” Molecular Systems Biology, vol. 17,
Sept. 2021.
N. Kassis-Chikhani, D. Edriss, L. Belec, A. Seve, L.
Courtellemont, H. Pe´re´, L. Hoc- queloux, S. Fafi-
Kremer, T. Prazuck, H. Mouquet, T. Bruel, E. Simon-
Lorie`re, F. A. Rey, and O. Schwartz, “Reduced
sensitivity of SARS-CoV-2 variant Delta to antibody
neutraliza- tion,” Nature, vol. 596, pp. 276–280, Aug.
2021.
R. C. Edgar, “[No title found],” BMC Bioinformatics, vol.
5, no. 1, p. 113, 2004.
R. McBride, M. van Zyl, and B. Fielding, “The Coronavirus
Nucleocapsid Is a Multifunc- tional Protein,” Viruses,
vol. 6, pp. 2991–3018, Aug. 2014.
S. Kumar, G. Stecher, M. Li, C. Knyaz, and K. Tamura,
“MEGA X: Molecular Evolutionary Genetics Analysis
across Computing Platforms,” Molecular Biology and
Evolution, vol. 35, pp. 1547–1549, June 2018.
S. Satarker and M. Nampoothiri, “Structural Proteins in
Severe Acute Respiratory Syndrome Coronavirus-2,”
Archives of Medical Research, vol. 51, pp. 482–491,
Aug. 2020.
V. M. Corman, O. Landt, M. Kaiser, R. Molenkamp, A.
Meijer, D. K. Chu, T. Bleicker,S. Bru¨nink, J.
Schneider, M. L. Schmidt, D. G. Mulders, B. L.
Haagmans, B. v. d. Veer, S. v. d. Brink, L. Wijsman, G.
Goderski, J.-L. Romette, J. Ellis, M. Zambon, M.
Peiris, H. Goossens, C. Reusken, M. P. Koopmans, and
C. Drosten, “Detection of 2019 novel coronavirus
(2019- nCoV) by real-time RT-PCR,”
Eurosurveillance, vol. 25, p. 2000045, Jan. 2020.
Publisher: European Centre for Disease Prevention and
Control.
X. Fang, J. Gao, H. Zheng, B. Li, L. Kong, Y. Zhang, W.
Wang, Y. Zeng, and L. Ye, “The membrane protein of
SARS-CoV suppresses NF-B activation,” Journal of
Medical Virology, vol. 79, pp. 1431–1439, Oct. 2007.
Y. Fan, K. Zhao, Z.-L. Shi, and P. Zhou, “Bat
Coronaviruses in China,” Viruses, vol. 11, p. 210, Mar.
2019.
Y. Huang, C. Yang, X.-f. Xu, W. Xu, and S.-w. Liu,
“Structural and functional properties of SARS-CoV-2
spike protein: potential antivirus drug development for
COVID-19,” Acta Pharmacologica Sinica, vol. 41, pp.
1141–1149, Sept. 2020.
Y. Jung, G.-S. Park, J. H. Moon, K. Ku, S.-H. Beak, C.-S.
Lee, S. Kim, E. C. Park, D. Park, J.-H. Lee, C. W.
Byeon, J. J. Lee, J.-S. Maeng, S.-J. Kim, S. I. Kim, B.-
T. Kim, M. J. Lee, and H. G. Kim, “Comparative
Analysis of Primer–Probe Sets for RT-qPCR of
COVID-19 Causative Virus (SARS-CoV-2),” ACS
Infectious Diseases, vol. 6, pp. 2513–2523, Sept. 2020.
Publisher: American Chemical Society.
The Phylogenetic Analysis of 5 SARS-CoV-2 Proteins’ Sequences in Relation to Time and Geographical Location
1173
