
Privacy-preserving Copy Number Variation Analysis with Homomorphic
Encryption
Hüseyin Demirci
a
and Gabriele Lenzini
b
Interdisciplinary Center for Security Reliability and Trust (SnT), University of Luxembourg,
6, avenue de la Fonte, L-4364 Esch-sur-Alzette, Luxembourg
Keywords:
Privacy-preserving Genomic Data Processing, Copy Number Variation, Homomorphic Encryption, Applied
Cryptography.
Abstract:
Innovative pharma-genomics and personalized medicine services are now possible thanks to the availability
for processing and analysis of a large amount of genomic data. Operating on such databases, is possible
to test for predisposition to diseases by searching for genomic variants on whole genomes as well as on
exomes, which are collections of protein coding regions called exons. Genomic data are therefore shared
amongst research institutes, public/private operators, and third parties, creating issues of privacy, ethics, and
data protection because genome data are strictly personal and identifying. To prevent damages that could
follow a data breach—a likely threat nowadays—and to be compliant with current data protection regulations,
genomic data files should be encrypted, and the data processing algorithms should be privacy-preserving.
Such a migration is not always feasible: not all operations can be implemented straightforwardly to be privacy-
preserving; a privacy-preserving version of an algorithm may not be as accurate for the purpose of biomedical
analysis as the original; or the privacy-preserving version may not scale up when applied to genomic data
processing because of inefficiency in computation time. In this work, we demonstrate that at least for a well-
known genomic data procedure for the analysis of copy number variants called copy number variations (CNV)
a privacy-preserving analysis is possible and feasible. Our algorithm relies on Homomorphic Encryption, a
cryptographic technique to perform calculations directly on the encrypted data. We test our implementation for
performance and reliability, giving evidence that it is practical to study copy number variations and preserve
genomic data privacy. Our proof-of-concept application successfully and efficiently searches for a patient’s
somatic copy number variation changes by comparing the patient gene coverage in the whole exome with a
healthy control exome coverage. Since all the genomics data are securely encrypted, the data remain protected
even if they are transmitted or shared via an insecure environment like a public cloud. Being this the first study
for privacy-preserving copy number variation analysis, we demonstrate the potential of recent Homomorphic
Encryption tools in genomic applications.
1 INTRODUCTION
Thanks to the technological revolution brought up by
Next Generation Sequencing (NGS), which makes
possible to sequence a human genome in hours and
for a few hundred USD, national large-scale projects
(e.g., the 100,000 Genomes Project
1
) and private ini-
tiatives can now afford sequencing the genomes of
hundreds of thousand of individuals. Large digital
genomic databases of sequenced raw data and other
genomic data (i.e., .bam files) are being created for
a
https://orcid.org/0000-0002-0538-2074
b
https://orcid.org/0000-0001-8229-327
1
https://www.genomicsengland.co.uk/ (11/2021)
present and future data analysis and research.
The availability of genomic databases is already
paving the way for innovative genomic medicine,
for instance personalized immunotherapeutic meth-
ods for cancer treatment (Kakimi et al., 2017), with
unquestionable benefits in terms of quality of future
treatments and healthcare. But, storing and process-
ing genomic data also raises serious concerns about
privacy rights: genomic data contains extremely sen-
sitive and personal information, for instance about the
owner’s, his/her relative’s, and future kin’s suscepti-
bility and predisposition to specific diseases such as
Alzheimer’s, schizophrenia, and cancer. This is not an
hypothetical threat: cases of misuse of genomic data
for individual discrimination have already reported
Demirci, H. and Lenzini, G.
Privacy-preserving Copy Number Variation Analysis with Homomorphic Encryption.
DOI: 10.5220/0011012400003123
In Proceedings of the 15th Inter national Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2022) - Volume 5: HEALTHINF, pages 821-831
ISBN: 978-989-758-552-4; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
821

(Naveed et al., 2015), showing that an extensive avail-
ability of genomic databases can have serious ethical
and legal concerns.
Several methods have been proposed and stud-
ied for genomic data protection. The most com-
mon and simplistic is pseudonymization i.e., remov-
ing from a genome data file any reference that could
link it back to its owner. For genomic data protection,
pseudonymization has been proved to be ineffective
(Gymrek et al., 2013). Genomic data are rich in in-
formation, and retrieve to whom a piece of genomic
data belong is easily done by triangulating data.
Another way to protect genomic data is to obfus-
cate (i.e., filter out) private sensitive sequences of nu-
cleotids from the raw data (Decouchant et al., 2018).
The idea is worth, but filtering information affects
the quality of processing, even if the filtering is done
selectively and in small amount. Besides, changing
well-established NGS protocols and machines require
time and effort, for instance due to the definition of
new standards, which is delaying the availability of
this technology.
Yet another option is to encrypt the genomic data
before they are stored. This strategy should at least
protect the data in case data leakage and as such,
promises to be compliant with legal requirements in
directives such as the Europe’s General Data Pro-
tection Regulation (GDPR) and the California Con-
sumer Privacy Act (CCPA). However, data base en-
cryption is insufficient to ensure a long-lasting pro-
tection if, for the purpose of processing, encrypted
data decrypted before use. Decrypted data are unpro-
tected. It is hard to control that they are not copied and
stored unencrypted, or re-transmitted in clear. The
risks of privacy violation remains high.
A better solution would be to run the analysis di-
rectly on the encrypted data without ever decrypt-
ing them. The practices is, at least in theory, possi-
ble thanks to secure data processing algorithms. Re-
searchers have explored this possibility and for spe-
cific procedures of genomic analysis, mostly about
small variants, namely Single-Nucleotide Polymor-
phisms and indels (i.e., small insertion - deletions).
The challenge is that producing a secure (i.e., privacy-
preserving) version of a specific data processing pro-
cedure requires redesigning the algorithm of analy-
sis by using only specific secure functionalities. A
privacy-preserving version of an algorithm with com-
parative and acceptable quality of analysis and per-
formance of the original version is not always possi-
ble. And even if it were possible a privacy-preserving
variant may remain inefficient, unable to scale up to
process large size data as those required for medical
purposes.
We study how to realize secure data processing
for an important class of genomic data procedures,
those aiming at the analysis of copy number varia-
tions (see Section 2.1). We assess the performances of
this new privacy-preserving processing, and we give
evident that a professional implementation can be ef-
ficient. To calculate copy number variation values on
encrypted genomic data, we use Homomorphic En-
cryption (HE) (see Section 2.4), a cryptographic tech-
nique that enables to operate directly and exclusively
on encrypted data and that produce results that are
still encrypted. By using HE, data processors never
get to know the genomic data they process, nor they
can make sense of the results of the processing unless
authorized to do so: both genomic data and the copy
number variation analysis can be safely outsourced to
third parties e.g., research laboratories specialized in
that type of analysis or a cloud service with sufficient
power of computation for the data processing.
Contribution. We design a proof-of-concept algo-
rithm for the detection of copy number variations that
uses HE operations, thus providing the first privacy-
preserving procedure for CNV analysis which relies
on this cryptographic technique. Our algorithm uses
basic homomorphic operations (addition, subtraction,
multiplication), for which an efficient implementation
exists in standard cryptographic software libraries,
making it possible to implement a prototype of the
procedure.
We test the quality of our privacy-preserving ver-
sion by looking for somatic copy number variation
changes in an exome sample
2
in comparison to ref-
erence exome samples used as control group. These
samples come from an existing Melanoma whole ex-
ome study (Magi et al., 2013).
The results of our privacy-preserving procedure
(once decrypted) are proven compatible with those re-
ported in the study. We also show that we can extend
our privacy-preserving procedure to calculate copy
number variation values for the whole genome and
not only for exomes. Our work reveals the potential
of using HE in the analysis of copy number variation
over an exome and potentially on the whole genome,
and that it is practical if we assume a convenient sam-
pling strategy.
Our proof-of-concept software and the data sets
we have used are on git
3
.
2
Exomes are collections of protein coding regions of the
genome, regions that are singularly called exons.
3
github.com/huseyindemirci44/privateCNVanalysis
Scale-IT-up 2022 - Workshop on Scaling-Up Health-IT
822

2 BACKGROUND
Before describing the core of our privacy-preserving
procedure, we remind a few basic facts on copy
number variation, variant data sets and analysis, and
Homomorphic Encryption (HE). We mention rele-
vant works on private genomic data processing that
use cryptographic techniques, and we comment their
use in copy number variation.
2.1 Copy Number Variation (CNV)
The Human Genome Project, the international sci-
entific research project that had been running from
1990 to 2003 and whose goal was determining the
base pairs of the human genome
4
, has revealed ab-
sence and replications of genetic material inside the
genome. These structural variants in the number of
copies of specific regions are the result of genomic
mutations, called deletions and duplications. They
cover large regions of the genome and involve several
genes.
Copy number variations are structural variants in
a person’s genome involving more than one thousand
bases. They differ from small variants such as in-
dels and repetitions that involves only a few bases. A
non-mutated genome region is expected to occour in
2 copies, respectively coming from the chromosomes
of the mother and of the father, but because of dele-
tion or duplication, the number of copy of the region
may respectively fall down to 1 or 0, or raise up to
3, 4, or more copies. These copy number variations
also change the number of genes and gene expression
levels inside copy number variation regions. Recent
studies claim that around 4.8% − 9.7% of the human
genome is copy number variation (Zarrei et al., 2015).
Generally copy number variations do not have
a direct phenotypic effect, but elevated copy num-
ber variations of particular genes have been shown
be associated with specific cancer types (Shlien
and Malkin, 2009; Shao et al., 2019). Changes
in copy number variation are correlated with in-
creased risk of developing psychiatric disorders such
as schizophrenia (Joober and Boksa, 2009; Marshall
et al., 2017); with major disease classes including
autoimmune, cardio-metabolic, oncologic, and neu-
ropsychiatric diseases (Li et al., 2020); and with
Mendelian and complex diseases such as Alzheimer
and autism (Zhang et al., 2009).
Needless to say, the interest to find copy number
variations and to understand how copy number varia-
tion affect to the phenotype is increasing.
4
An organism’s genome is its complete set of DNA.
2.2 CNV Analysis
It is a common practice to keep and maintain data
bases of genomic variants for research. For instance,
DECIPHER (Bragin et al., 2014) and “Database of
Genomic Variants” (MacDonald et al., 2014) stores
copy number variation data for both patient and
healthy individuals. Data were usually calculated us-
ing traditional methods such as fluorescence in situ
hybridization (FISH) and array comparative genomic
hybridization (aCGH), but nowadays computational
methods working on digital genomic data are pre-
ferred.
The detection of copy number variation is done
using specif algorithms and procedures, and we refer
to (Zhang et al., 2019) and (Moreno-Cabrera et al.,
2020) for a comprehensive discussion on them. In
this work, we refer to one of such methods, the Ex-
cavator tool (Magi et al., 2013), whose output values
are reported Table 1. They tell about the presence of
variations in a genome. Copy number variants can be
calculated using read depth of coverage information
(Yoon et al., 2009). For instance, in the Excavator
tool, the mean read count (EMRC) for each exon e in
a genomic region is calculated by the following for-
mula:
EMRC =
RC
L
where RC is the number of reads aligned in the region
and L is its size in base pairs. EMRC calculated from
the genome of an individual are then compared with
reference values calculated from control genomes, af-
ter a normalization step.
Comparing with control genomes is a common
method in structural variant analysis. It serves to filter
polymorphic variants and to increase statistical signif-
icance. Statistical methods, such as Hidden Markov
Models, are used to estimate the locations and prob-
ability of genomic events. Such a justified need to
analyze this class of genomics variants as well as
other variants would surely benefit from methods of
processing that are privacy-preserving and capable to
protect the personal sensitive health related informa-
tion contained in the genome of patients without com-
promising the quality of the analysis.
2.3 Private Genomic Data Processing
There has been several studies about using crypto-
graphic technique in ensuring privacy in genomic
data processing. Differential Privacy, tailored to ge-
nomic databases, has been tested to enable privacy-
preserving Genome Wide Association Study (GWAS)
applications (Simmons et al., 2016). Another tech-
nique, Secure Multiparty Computation, has been
Privacy-preserving Copy Number Variation Analysis with Homomorphic Encryption
823

Table 1: Copy Number Variation.
Copy Number Explanation Notation
Double Gain ≥ 2 extra copies are duplicated 2
One copy gain Extra 1 copy is obtained 1
Normal case 2 copies exist 0
Heterozygous deletion One of the copies is lost -1
Homozygous deletion There is no DNA sequence -2
tested in “trio exome analysis” for small cohorts of
patients, and demonstrated to be executable in sec-
onds whereas the remaining variant set of any patient
remained privately protected (Jagadeesh et al., 2017;
Akgün et al., 2020). These methods are not directly
applicable to copy number variation analysis, because
the detection of variants is generally out of their work-
ing pipeline, which focus on Single-Nucleotide Poly-
morphisms and short indels. Instead,Homomorphic
Encryption (HE) has been applied to realize a se-
cure exact logistic regression model for rare disease
GWAS studies (Wang et al., 2016), but to our best
knowledge at the moment of writing
5
, there is no spe-
cific work that studies how to protect genomic privacy
in copy number variation detection, nor one that pro-
poses using HE for this task. Once copy number vari-
ation values are calculated in a private manner and
stored (even in encrypted form) in a .vcf file, all the
cryptographic methods previously mentioned can be
employed to ensure privacy protection in further data
analysis, tests, and processing.
2.4 Homomorphic Encryption
This cryptographic technique can be considered as a
bridge (i.e., an homomorphism) between operations
on a plaintext (unencrypted) domain and operations
on an encrypted domain. This means that HE makes
it possible to perform computations directly on the en-
crypted. The output of the computation, which is en-
crypted, once decrypted, is the same as the output that
would have been produced if the computation were
performed on the unencrypted data.
For example, an additive HE scheme would ensure
that Enc
k
(d + d
0
) = Enc
k
(d) ⊕ Enc
k
(d
0
), where Enc
is the encryption function, k encryption key, ⊕ the
homomorphic addition, and d and d
0
pieces of data.
HE enables outsourcing the computation of per-
sonal data while ensuring privacy. A subject that has
encrypted some personal data by using its public key
can send it safely to an insecure party to get them pro-
cessed. The party processes the data without ever get-
ting access to the plain data and returns the encrypted
results back to the subject who is the only one who
5
June 2021
can decrypt them by using the private key. Even if the
data breach were breached on the party side, confiden-
tiality is ensured because the data cannot be decrypted
without the private key.
For reasons of space, we have to limit our account
of HE schemes. The reader can refer to (Acar et al.,
2018) for a comprehensive survey on their theory and
implementations, but for the scope of this work we
point out that there are different types of HE schemes.
The most common ones are the following:
• Partially Homomorphic Encryption. They pre-
serve the homomorphism only for computations
using one operation, for example only addition,
or only multiplication.
• Somewhat Homomorphic Encryption. They pre-
serve homomorphism for bounded computations,
for instance computation using a specific number
of additions and multiplications.
• Fully Homomorphic Encryption (FHE). They are
schemes that preserve the homomorphism for ar-
bitrary computations, for instance, computations
with an arbitrary number of additions and multi-
plications.
The latter is the most general, and a proof for its ex-
istence for arbitrary computation using addition and
multiplication has been presented in 2009 by Gentry
who used mathematical lattices for the purpose (Gen-
try, 2009). Most of the HE schemes of today also
rely on solutions for hard-to-solve computational al-
gebraic problems over lattices, known as Ring Learn-
ing with Errors (Regev, 2009). Recently, libraries
such as Microsoft’s SEAL
6
and IBM’s Fully Homo-
morphic Encryption Toolkit For Linux
7
have been
made available for developing HE applications.
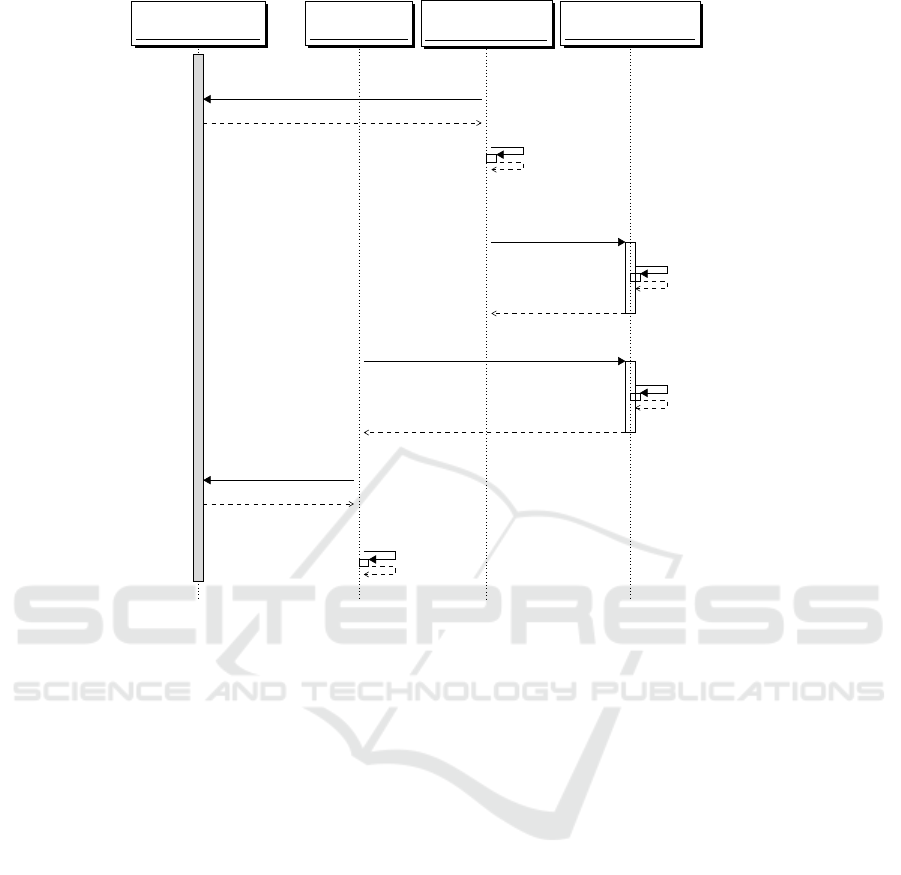
3 REFERENCE SCENARIO
To contextualize the copy number variation (CNV)
analysis with HE, let us refer to the following situ-
ation. A patient, who had his genome sequenced and
6
https://github.com/microsoft/SEAL
7
https://github.com/IBM/fhe-toolkit-linux
Scale-IT-up 2022 - Workshop on Scaling-Up Health-IT
824
Trusted Party (TP)
(pk,sk)
Medical Unit
(MU)
Patient P
0
Cohort Patients
~
P
Storage Processing
Unit (SPU)
1. Req(PubKey)
pk
Encrypt
g
0
,~g
Enc
pk
(g
0
), Enc
pk
(~g)
2. Enc
pk
(g
0
), Enc
pk
(~g)
Store
encrypted data
3. Req(CNV)
Calculate Secure CNV
on Enc
pk
(g
0
), Enc
pk
(~g)
Enc
pk
(CNV(g
0
,~g))
4. Enc
pk
(CNV(g
0
,~g))
5. Req(PrivKey)
sk
Decrypt Enc
pk
(CNV(g
0
,~g))
using sk
CNV(g
0
,~g)
Figure 1: Message flow for a secure processing in our scenario.
genomic data coverage files stored in a biobank, con-
sults a doctor. The doctor suggests a test which re-
quires identifying copy number variants because he
intends to investigate changes in the tumor suppressor
genes like TP53 for a possible drug resistance anal-
ysis. This could ideally be carried out by the same
biobank by comparing the patient’s tumor sequence
coverage data with those other patients used as a con-
trol set.
Since both the patient’s and other patients’ ge-
nomic data, in this example the sequence coverage
files, are personal data, such data are stored encrypted
by the biobank.
Figure 1 shows a possible secure processing pro-
tocol. The message exchange involves the follow-
ing agents: a Trusted Party (TP), usually an authority
such as the Public Health System Institute or a na-
tional Certified Institution, which is in charge of cre-
ating a pair of encryption-decryption keys (pk, sk), re-
spectively, public and private meant to be used in sup-
port to secure genomic processing; the Medical Unit
(MU), usually a doctor or a researcher in the need of
processing genomic data; the Storage and Processing
Unit (SPU), which is generally represented by a pub-
lic cloud service for instance run by a biobank; the
patients, which are the data subjects of the genomic
data, which herein we assume to be a particular pa-
tient’s genomic data (P
0
and g
0
), and the genomic data
files of a cohort of reference patients (
~
P and ~g).
Figure 1 protocol exchange is as follows: Before
storing their genomic data on the clouds, the patients
(P
0
, as well as
~
P), request (message 1) and obtain the
public key pk from TP, using which they encrypt their
genomic data, respectively g
0
and ~g. The encrypted
data, resp. Enc
pk
(g
0
), Enc
pk
(~g), are send to the SPU
(message 2), which stores them for future uses.
When a MU, having agreed with patient P
0
to
make a CNV analysis for which it needs the patient’s
genome g
0
and/or those of the cohort (~g), makes a re-
quest for the analysis to the SPU (message 3). The
SPU performs on behalf of MU the calculation by
running the secure homomorphic encryption version
of copy number variation algorithm (see later, Algo-
rithm 1) on the encrypted genomic data previously
stored. The SPU sends back to the MU the encrypted
outcome of the analysis, Enc
pk
(CNV(g
0
,~g)), which
cannot yet be decrypted. MU requests for and ob-
tains the decryption key from the TP (message 6), af-
ter which MU gets to know the result of the analysis.
None of the agents but the data subjects even know
the patients’ personal genomic data in cleartext. It
is important to stress that the protocol in Figure 1
Privacy-preserving Copy Number Variation Analysis with Homomorphic Encryption
825

does not protect against active intruders, such as an
SPU that tries to fool TP into getting the private key.
Additional measures need to be in place for reach-
ing higher levels of security, which is a goal out of
the scope of this work. The protocol is meant to en-
sure data privacy against a passive but curious SPU,
for which the use of privacy-preserving copy number
variation is an adequate measure.
4 A PRIVACY-PRESERVING CNV
We now present our implementation of the secure
CNV detection that relies on HE. The pseudocode
is reported in Algorithm 1.
For simplicity of exposition, we assume that all
the computations are performed locally in one ma-
chine instead of being distributed as they should be
according to Figure 1. Consequence of this simpli-
fication is that we do distinguish between different
roles, not we show the interroles communication. The
feasibility and performance benchmarks that we ob-
tain from the experiment remain valid because we are
not interested in measuring the overhead due to the
communication but only the impact of the HE com-
putation for a CNV analysis on genomic data.
The procedure PPCNV is inspired by the pro-
cedure we described in Section 2. It takes in input
the coverage files
8
for patient and control cases, re-
spectively, CovPatient and CovControl, and the list of
genes Genelist to point out the regions to be analyzed.
It returns the list CNV of copy number predictions for
the genes in Genelist. The procedure refers to certain
threshold parameters (th
i
in the pseudocode), which
represent the boundary conditions to have 0, 1, 2, 3, 4
or more copies of DNA parts.
The rationale of procedure PPCNV is as follows
(we assume basic knowledge about coverage files, ex-
omes, and genes): the patient’s gene coverage file,
which contains the number of occurrences of each
gene on the different reads (i.e., overlapping frag-
ments) of the patient’s genome after sequencing, is
compared with a control coverage file of an healthy
individual. The number of copy variants for a gene
g is determined looking at the difference between the
coverage files. For instance if the difference is zero,
then there is no variants; if it is bigger than 2 there
are more than 2 copy variants. Intermediate values
are determined reasoning on the value of the differ-
ence: if it is bigger then 1.5, there are quite likely 2
copy variants, whereas if it is bigger then 0.5, there is
quite likely 1 copy variation. We use a window of five
8
Coverage files contain information regarding the
base/read coverage of each sequence in an assembly file.
overlapping consecutive gene regions, and we take the
value that occurs in the majority of the cases as the
final copy variant value. The key point of PPCNV
is to implement this ideas using homomorphic opera-
tions, namely ⊗ and ⊕ (and therefore ). There is no
homomorphic operator that can be used to determine
whether e.g., (x−y) > 1.5. The idea is then to rewrite
the expression as x − (1.5 × y), which can be calcu-
lated homomorphically. That done, we can encrypt
the coverage files and calculate x (1.5 ⊗ y) for all
genes, keeping the sensitive coverage files protected.
Algorithm 1: Privacy-preserving CNV Procedure.
PPCNV(CovPatient,CovControl, Genelist)
1
2 // SPU calculation
3 forall g ∈ Genelist
4 for i = 0 to 4 do
5 c
g
i
:= Enc
pk
(th
i
⊗ CovControl
g
);
6 for i = 0 to 3 do
7 d
g
i
:= c
g
0
c
g
i+1
8 return (d
g
0
,d
g
1
,d
g
2
,d
g
3
)
9
10 // MU calculation
11 forall g ∈ Genelist
12 for i = 0 to 3 do
13 d
g
i
:= Dec
sk
(d
g
i
)
14 n
g
:= 0 ;
15 if d
g
0
> 0 then n
g
:= 2
16 else if d
g
1
> 0 then n
g
:= 1
17 else if d
g
2
< 0 then n
g
:= −1
18 else if d
g
3
< 0 then n
g
:= −2;
19 n
x
:=
20 maxCount({n
g
,n
g+1
,n
g+2
,n
g+3
,n
g+4
})
21 CNV
g
:= n
x
22 return CNV
g
In Algorithm 1 Enc and Dec represent HE en-
cryption and decryption operations with the public
and private keys. Values th
1
,th
2
,th
3
,th
4
are the HE
threshold parameters where th
1
≈ 2., th
2
≈ 1.5, th
3
≈
0.5 and th
4
≈ 0. The threshold numbers represent the
boundary conditions to have 0, 1, 2, 3, 4 or more
copies of DNA parts. After obtaining the initial copy
number variant predictions, we select the prediction
value that occurs with maximal frequency (i.e., func-
tion maxCount ) over a non-overlapping window of
length 5 to filter out instant fluctuations since the copy
number variation values do not change frequently.
The most frequent element in the window is estimated
as the copy number variation value for that window.
In Algorithm 1, we made another simplification,
this time with consequences. The final calculation of
copy number variation values is estimated after cer-
Scale-IT-up 2022 - Workshop on Scaling-Up Health-IT
826

tain intermediate values, d
g
s in the code, have been
decrypted (step 11–13). This means that the SPU has
to send the d
g
values back to the MU, and it will MU
that finalizes the calculation.
This simplification is motivated because imple-
menting homorphically the test ‘> 0’ (lines 16–18)
is not straightforward and it would introduce a com-
putational burden that seems unnecessary. The ge-
nomic data to protect are the coverage files, while the
encrypted values d
g
, once sent back to the MU, who
decrypts them, do not disclose, to MU, more informa-
tion then the values CNV
g
, which MU gets to know
eventually.
The current implementation runs efficiently be-
cause we implemented HE encryption and decryption
using natif3 operations for vectors. The comparison
for the list of whole exome genes is calculated using
4 vector encryption-decryption pairs and the required
intersections conveniently as described in Algorithm
1. In this way, we could label all the gene coverages to
a CNV value using the threshold parameters. The ac-
tual values of the parameters can be adjusted accord-
ing to the coverage settings, tumor/normal sample ra-
tio, including the normalization step for the samples.
5 EXPERIMENTAL SETTING,
DATA SETS, AND LIBRARIES
To test the feasibility of this CNV query using HE we
have set up the following experiment setting. We con-
sidered a publicly available Melanoma tumor sample
data and compared it with a healthy control individ-
ual. We have calculated the mean read count (GMRC)
per gene as follows:
GMRC =
RC
L
where RC is the number of reads aligned in the gene,
L is the size of the gene in base pairs. We use
GMRC values to make a comparison for case and
control cases. First, we compare GMRC values for
a single gene such as TP53 or PTEN. Later we have
used the same comparison query for a list consist-
ing of 100 genes, representing a gene panel. Then
we have carried out analysis over the chromosome
level. We have used the whole list of 1228 genes in
Chromosome 9. This chromosome is chosen since
it contains CDKN2A gene which is highly damaged
in Melanoma cancer (Pfarr et al., 2016). Finally,
we have conducted this experiment on the whole set
of Refseq curated list of genes, consisting of 23.882
genes out of 31.848 locations where both the samples
have coverage greater than 5 (Supplementary Files:
geneslist.txt, allgeneshealthy.csv, allgenespatient.csv,
genes_Ch9_healthy.csv genes_Ch9_patient.csv ). We
have measured the performance of the encryption and
decryption processes for these queries. We have com-
pared our CNV results for Chromosome 9 with the
original study (Magi et al., 2013) to analyze the accu-
racy of the secure CNV detection algorithm.
The data we have used in this paper has been
taken from the samples that has been used in the
Excavator CNV detection tool (Magi et al., 2013).
The data consists of a tumor whole exome sequence
from a Melanoma patient (Melanoma
01
) and a con-
trol healthy individual (Sample
01
). We have uploaded
the .bam files of the samples with accession num-
ber ERR174231 and ERR174237 which were aligned
according to Genome Reference Consortium Human
Build 37 (GRCh37, hg19) from the Sequence Read
Archive (SRA) database (Leinonen et al., 2010) to
the Galaxy web platform. We have used the pub-
lic server usegalaxy.org (Afgan et al., 2018) to calcu-
late the coverage values with .bed tools. Then using
these base read coverage values, we calculate the read
counts per gene (GMRC) as described above, which
provide the input of Algorithm 1.
The HE operations throughout this work are ex-
ecuted with the help of Pyfhel: PYthon For HE Li-
braries
9
which uses the SEAL (Chen et al., 2017) li-
brary as a backend. Pyfhel provides vector and frac-
tion encryption/decryption capabilities which were
especially helpful while performing operations on the
list of genes. We note that the available implemen-
tations of HE only allow simple operations such as
addition, subtraction, and multiplications.
6 RESULTS
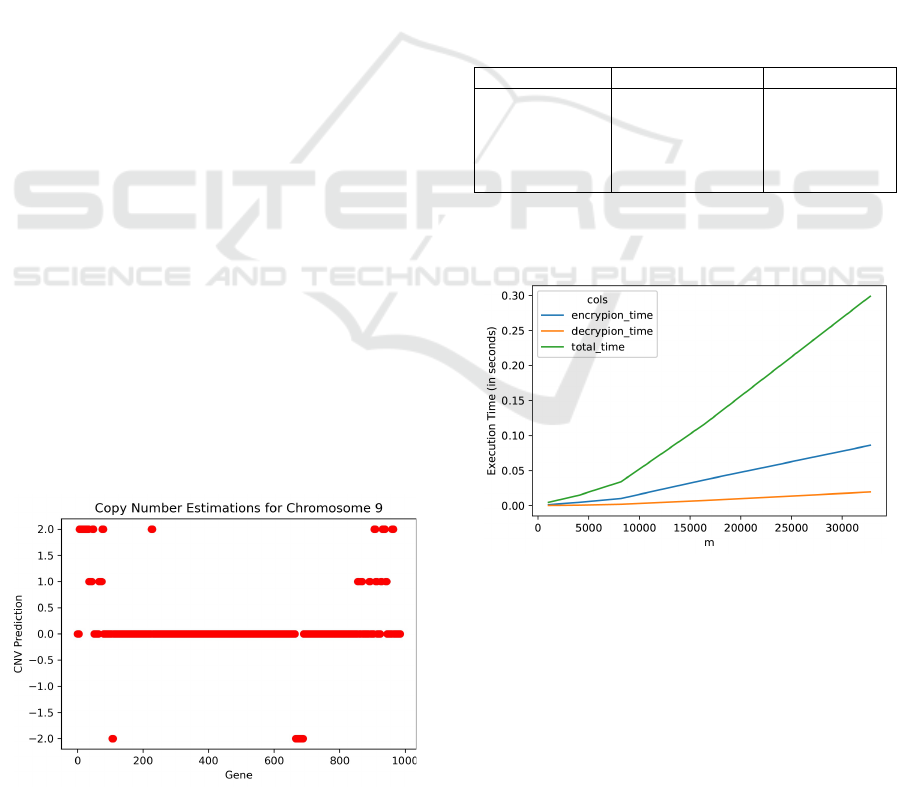
Using Algorithm 1, we have compared the coverage
values gathered from two .bam files. Figure 2 depicts
the copy number variant estimations for Chromosome
9 obtained by comparing the Melanoma tumor file
with the healthy sample file which indicates double
copy gains at the beginning and end of Chromosome
9, and two double deletion regions. We also detect
one copy gains at certain points. We check these re-
sults against the CNV values reported in the original
study (Magi et al., 2013). Among these variants, 29 of
the 30 double deletion genes are the same with (Magi
et al., 2013). Our algorithm has predicted the CNV
region a little larger so that we have the MTAP gene
in the boundary. In particular we are able to detect the
double loss of CNV in the gene CDKN2A which is as-
9
https://github.com/ibarrond/Pyfhel
Privacy-preserving Copy Number Variation Analysis with Homomorphic Encryption
827
sociated with Melanoma cancer. For double copy gain
genes, 22 of the 65 do not appear in the Excavator list.
This comes from the fact that Excavator algorithm did
not report the variations at the end of Chromosome
9. Both works predicted no one copy losses wher-
ever our algorithm detected 50 one copy gains where
19 of them were reported as double gain in Excava-
tor list. Excavator did not identify any one copy gains
or losses. Table 2 presents the ratio of genes that are
consistent with the results of the (Magi et al., 2013)
tool.
The whole list of genes and their estimated CNVs
for Chromosome 9 are presented in the Supplemen-
tary File (Chromosome9_Estimated_CNV.csv). It is
an expected phenomena that two CNV detection al-
gorithms may slightly differ since they have different
sensitivity to parameters such as depth of the sequenc-
ing and they use different statistical models (Zhang
et al., 2019). Nevertheless, the comparison results
demonstrate the usability of a HE based CNV predic-
tion. We emphasize that our aim is not to improve the
performance of CNV detection but to demonstrate the
feasibility of a secure version of CNV analysis using
existing HE framework.
Computational Performances. The time perfor-
mance of the HE experiment is provided in Table 3.
These experiments are carried out with a laptop with
i-7 1.8- 2.3 GHz. CPU and 16 GB RAM with 64
bit Windows Enterprise operating system. We used
Python version 3.7.4 on the Spyder Environment. We
note that time performance depends on the HE pa-
rameters. For a single gene value the calculations can
be executed using Pyfhels’s homomorphic operations
for fraction. For larger gene lists, vector operations
can be used. To have a decryption without noise, the
vector parameter defined by m must be larger than
our query list. Figure 3 represents the performance
of the encryption, decryption and total time processes
Figure 2: Copy Number Estimations for Chromosome 9.
which are collected while encrypting a list of 1000
genes with respect to the HE parameter m. The graph
demonstrates a linear increase for encryption and de-
cryption times according to the parameter m. For a
fixed vector size m, the execution times do not change
with the length of the gene list since actually the al-
gorithm implements encryption and decryption oper-
ations along the vector size. In the worst case where
we choose the vector size m = 32.768 to ensure the
inclusion of the whole exome gene list in the encryp-
tion array, the results are still in the order of decisec-
onds. In Table 3 only the time performance of the
HE operations are demonstrated. The preprocess re-
quired to obtain the read count tables have not been
considered since, this is a standard step in any CNV
analysis without an encryption. Our aim is to measure
the workload of the computations executed in the en-
crypted domain.
Table 2: Comparison of CNVs for Chromosome 9 with
(Magi et al., 2013).
CNV Detected Number of genes Overlap ratio
∗
2 65 0.66
1 50 0.38
∗∗
0 841 0.90
-1 0 1
-2 30 0.96
∗
The ratio of genes that exist in (Magi et al., 2013)
∗∗
19 of the 50 Double Gain genes are found in the in One Copy
list of (Magi et al., 2013).
Figure 3: Time performance according to the encryption
parameter m.
7 DISCUSSION
What we have exposed so far shows that is feasible to
code a CNV analysis algorithm using HE, that repre-
sents a privacy-preserving version of CNV detection.
This task is greatly simplified thanks to software li-
braries that offer reliable and optimized implemen-
tations of HE operations such as vector encryption
Scale-IT-up 2022 - Workshop on Scaling-Up Health-IT
828

Table 3: Time performance of the HE experiments. The Execution Time includes the Encryption + Decryption Time
Analysis Region Number of
Locations
HE parameters Execution
Time (Sec.)
Single Gene 1 p = 65.537, m = 1.024 0.0005
Gene Panel 100 p = 65.537, m = 1.024 0.0035
Chromosome 9 1.289 p = 65.537, m = 2.048 0.0088
Whole Exome 23.882 p = 65.537, m = 32.768 0.3448
Whole Genome
∗∗
320.00 p = 65.537, m = 32.768 4.7972
∗∗
Estimated Time from whole exome study
and decryption, and that they already solve otherwise
time-consuming and tricky technical issues e.g., boot-
strapping strategies to control noise in the scheme.
Our experiments show that the execution is effi-
cient, that is within seconds. We estimate that it re-
mains efficient when extended to processing a whole
genome.
We also demonstrated that this privacy-preserving
version of the algorithm performs a meaningful anal-
ysis of copy number variants. This has been demon-
strated initially by comparing two coverage files for
certain genomic positions or a gene panel, and then
extending the approach to process the whole exome
level. We speculate that it can be further extended
to a whole genome analysis, if we apply a cover-
age comparison strategy. For instance, 10% of the
whole genome can be covered with a window size of
1000 bases, which will result around 320.000 cov-
erage regions to compare which is of order 10-15
times larger magnitude compared to the whole ex-
ome study. Since copy number variation values do
not change frequently through the genome, we may
identify the values with good precision by sampling
10% of the genome. If this speculation is correct, the
whole genome CNV analysis can still be conducted in
seconds.
In our use case, we have taken a healthy exome
sequence as control to simulate somatic CNV detec-
tion. For a standard CNV analysis, the number of
samples can be increased to improve statistical signif-
icance. In this case, the execution time will increase
linearly with the number of samples. Notably, this
step is completely parallelizable where we can use
the computational power of the public cloud environ-
ment. Since the encryption-decryption processes are
fast, the total time performance still remains feasible
for real time applications.
An alternative strategy to direct comparison of
genomes is calculating the mean of the copy num-
ber variation values over the control samples as a base
line and use this base line as a reference for CNV de-
tection. The required averaging computation can be
done in the encrypted domain and needs to be exe-
cuted only once in the offline mode. Then comparing
each genome coverage with this average control base-
line will have the similar performance with this work.
The results demonstrate that the existing libraries
of HE enable real time analysis for small files like
coverage or .vcf files. However, a complete genomic
analysis pipeline requires to perform operations on
raw file types such as .sam, .bam or .fastq. At
the moment performing tasks like genome alignment
on encrypted data is not feasible since there are yet
no efficient algorithms for operations like compari-
son of two strings in the encrypted domain. More-
over, whenever there are conditional statements, the
HE implementation slows down substantially and the
data size expands as a result of re-linearization pro-
cess in encryption. Therefore, it is crucial to define an
algorithm with the most basic operations.
Security Assumptions and Parameters. In the
proposed protocol we have deployed a Trusted Party
for the generation of encryption and decryption keys.
TP as a certified and controlled institute, could access
genomic data but has not storage and processing ca-
pabilities. These tasks are executed at the storage and
processing unit, SPU. The SPU is not reliable. A pas-
sive malicious adversary who has somehow access to
the SPU (the admin of SPU, for instance), can not ob-
tain sensitive data since the security of the HE is guar-
anteed by the Public Key encryption system. The im-
plemented HE algorithm in Pyfhel library is the BGV
somewhat homomorphic encryption scheme without
any bootstrapping
10
. This method also depends on
the Learning with Errors over Rings (RLWE) prob-
lem and we refer the interested reader to the paper
(Brakerski et al., 2014) for mathematical details of the
algorithm. We have chosen a security level equivalent
to 128-bit symmetric (AES) encryption according to
the suggestions in (Albrecht et al., 2018). The pa-
rameter p is the integer modulus and m is the vector
size where the Ring operations are defined. The pri-
vate and public keys of the Homomorphic Encryption
system are generated according to the specifications
of the scheme, where public key is a secret Ring el-
10
Personal communication with the developers.
Privacy-preserving Copy Number Variation Analysis with Homomorphic Encryption
829

ement and private key is a pair of two Ring elements
which satisfy certain criteria, respectively.
We note that the requirement of a Trusted Party is
inevitable for this kind of study, since for the deriva-
tion of the genome sequence at least, there needs to be
a certified trusted authority. We note that previous re-
lated work also followed this assumption (see (Wang
et al., 2016; Ayday et al., 2013)).
Limitations. We limited our HE to simple arith-
metic operations in our redesigned CNV algorithm,
leaving to the MU (i.e., the doctor) the task to run in-
equality tests on the plain data. We argued that this
choice does not affect security, but it would be inter-
esting to measure the performance of a full HE ver-
sion of CNV. We leave it for future work.
An obvious limitation of this work is the number
of cases analyzed. As we intended to prove that a
copy number variation analysis using HE is feasible,
we chose as example the analysis of a single exome.
We plan to extend this work to several exoms and
whole genome samples to have a better evaluation of
accuracy and fine tuning of the threshold parameters
in future work.
Another issue is the normalization of sequenc-
ing data coming from various runs and technologies.
Comparison of data obtained from different sequenc-
ing technologies or parameters is a challenging pro-
cess. Tumor-normal sampling ratio is also of critical
importance while working with tumor data. A care-
ful normalization is required before data analysis for
accurate CNV results.
8 CONCLUSIONS
In this work we have presented a proof-of-concept
study for the estimation of copy number variation val-
ues by implementing a secure coverage comparison
using Homomorphic Encryption (HE).
We are able to calculate the copy number values
for a predefined list of genes privately without reveal-
ing personal genomic information. We have worked
on a somatic diagnosis use case with a Melanoma
whole exome data and compared the copy number
variation results with the original study (Magi et al.,
2013). Our results demonstrate the practicality and
accuracy of a privacy-preserving copy number varia-
tion (CNV) analysis at the whole exome and whole
genome levels.
As CNV analysis takes a broader deployment in
genomic pipelines, making it possible to perform in
a privacy preserving manner in compliance with data
protection regulations is becoming important. This
study is the first work focusing on this important class
of variants which have been neglected up to now.
As a future work, we plan to apply the proposed
CNV method to a set of whole genome sequences.
This will satisfy a better evaluation for the validity of
the sampling strategy and its precision on large whole
genome cohorts and more accurate determination of
the algorithm parameters.
Another upcoming line of research is the use of
HE for privacy-preserving .vcf queries to execute
applications such as private rare mutation discovery,
variant prioritization for causes of disease, and dis-
ease predisposition calculation. Although there exist
individual examples, there is still work to do for de-
veloping more efficient and unified approaches. Re-
cent HE libraries enable various types of genomic ap-
plications which can now be executed in real time hin-
dering mathematical and implementation difficulties
and opening the door for the age of privacy-preserving
personalized medicine.
ACKNOWLEDGEMENTS
This work has been supported by the EU 956562,
MSCA-ITN-2020 - Innovative Training Networks,
“Legality Attentive Data Scientists” (LeADS). The
authors wish to thank Dr. Patrick May for valuable
discussions on the topic of copy number variation.
REFERENCES
Acar, A., Aksu, H., Uluagac, A. S., and Conti, M. (2018).
A survey on homomorphic encryption schemes: The-
ory and implementation. ACM Computing Surveys
(CSUR), 51(4):1–35.
Afgan, E., Baker, D., Batut, B., Van Den Beek, M., Bou-
vier, D.,
ˇ
Cech, M., Chilton, J., Clements, D., Coraor,
N., Grüning, B. A., et al. (2018). The Galaxy platform
for accessible, reproducible and collaborative biomed-
ical analyses: 2018 update. Nucleic acids research,
46(W1):W537–W544.
Akgün, M., Ünal, A. B., Ergüner, B., Pfeifer, N., and
Kohlbacher, O. (2020). Identifying disease-causing
mutations with privacy protection. Bioinformatics.
Albrecht, M., Chase, M., Chen, H., Ding, J., Goldwasser,
S., Gorbunov, S., Halevi, S., Hoffstein, J., Laine,
K., Lauter, K., Lokam, S., Micciancio, D., Moody,
D., Morrison, T., Sahai, A., and Vaikuntanathan, V.
(2018). Homomorphic Encryption Security Stan-
dard. Technical report, HomomorphicEncryption.org,
Toronto, Canada.
Ayday, E., Raisaro, J. L., McLaren, P. J., Fellay, J., and
Hubaux, J.-P. (2013). Privacy-preserving computation
Scale-IT-up 2022 - Workshop on Scaling-Up Health-IT
830

of disease risk by using genomic, clinical, and envi-
ronmental data. In 2013 USENIX Workshop on Health
Information Technologies (HealthTech 13).
Bragin, E., Chatzimichali, E. A., Wright, C. F., Hurles,
M. E., Firth, H. V., Bevan, A. P., and Swaminathan,
G. J. (2014). DECIPHER: database for the interpre-
tation of phenotype-linked plausibly pathogenic se-
quence and copy-number variation. Nucleic acids re-
search, 42(D1):D993–D1000.
Brakerski, Z., Gentry, C., and Vaikuntanathan, V. (2014).
(leveled) fully homomorphic encryption without boot-
strapping. ACM Transactions on Computation Theory
(TOCT), 6(3):1–36.
Chen, H., Laine, K., and Player, R. (2017). Simple en-
crypted arithmetic library-SEAL v2. 1. In Inter-
national Conference on Financial Cryptography and
Data Security, pages 3–18. Springer.
Decouchant, J., Fernandes, M., Völp, M., Couto, F. M.,
and Esteves-Veríssimo, P. (2018). Accurate filtering
of privacy-sensitive information in raw genomic data.
Journal of Biomedical Informatics, 82:1–12.
Gentry, C. (2009). Fully homomorphic encryption using
ideal lattices. In Proceedings of the forty-first annual
ACM symposium on Theory of computing, pages 169–
178.
Gymrek, M., McGuire, A. L., Golan, D., Halperin, E., and
Erlich, Y. (2013). Identifying Personal Genomes by
Surname Inference. Science, 339(6117):321–324.
Jagadeesh, K. A., Wu, D. J., Birgmeier, J. A., Boneh, D.,
and Bejerano, G. (2017). Deriving genomic diag-
noses without revealing patient genomes. Science,
357(6352):692–695.
Joober, R. and Boksa, P. (2009). A new wave in the genet-
ics of psychiatric disorders: the copy number variant
tsunami. Journal of psychiatry & neuroscience: JPN,
34(1):55.
Kakimi, K., Karasaki, T., Matsushita, H., and Sugie, T.
(2017). Advances in personalized cancer immunother-
apy. Breast cancer (Tokyo, Japan), 24(1):16—24.
Leinonen, R., Sugawara, H., Shumway, M., and Collabora-
tion, I. N. S. D. (2010). The sequence read archive.
Nucleic acids research, 39(suppl_1):D19–D21.
Li, Y. R., Glessner, J. T., Coe, B. P., Li, J., Mohebnasab,
M., Chang, X., Connolly, J., Kao, C., Wei, Z., Brad-
field, J., et al. (2020). Rare copy number variants in
over 100,000 European ancestry subjects reveal mul-
tiple disease associations. Nature communications,
11(1):1–9.
MacDonald, J. R., Ziman, R., Yuen, R. K., Feuk, L.,
and Scherer, S. W. (2014). The database of ge-
nomic variants: a curated collection of structural vari-
ation in the human genome. Nucleic acids research,
42(D1):D986–D992.
Magi, A., Tattini, L., Cifola, I., D’Aurizio, R., Benelli,
M., Mangano, E., Battaglia, C., Bonora, E., Kurg, A.,
Seri, M., et al. (2013). EXCAVATOR: detecting copy
number variants from whole-exome sequencing data.
Genome biology, 14(10):R120.
Marshall, C. R., Howrigan, D. P., Merico, D., Thiruvahin-
drapuram, B., Wu, W., Greer, D. S., Antaki, D.,
Shetty, A., Holmans, P. A., Pinto, D., et al. (2017).
Contribution of copy number variants to schizophre-
nia from a genome-wide study of 41,321 subjects. Na-
ture genetics, 49(1):27–35.
Moreno-Cabrera, J. M., Del Valle, J., Castellanos, E., Feli-
ubadaló, L., Pineda, M., Brunet, J., Serra, E., Capellà,
G., Lázaro, C., and Gel, B. (2020). Evaluation of CNV
detection tools for NGS panel data in genetic diagnos-
tics. European Journal of Human Genetics, pages 1–
11.
Naveed, M., Ayday, E., Clayton, E. W., Fellay, J., Gunter,
C. A., Hubaux, J.-P., Malin, B. A., and Wang, X.
(2015). Privacy in the Genomic Era. ACM Comput.
Surv., 48(1).
Pfarr, N., Penzel, R., Klauschen, F., Heim, D., Brandt,
R., Kazdal, D., Jesinghaus, M., Herpel, E., Schir-
macher, P., Warth, A., et al. (2016). Copy number
changes of clinically actionable genes in melanoma,
non-small cell lung cancer and colorectal cancer—A
survey across 822 routine diagnostic cases. Genes,
Chromosomes and Cancer, 55(11):821–833.
Regev, O. (2009). On lattices, learning with errors, random
linear codes, and cryptography. Journal of the ACM
(JACM), 56(6):1–40.
Shao, X., Lv, N., Liao, J., Long, J., Xue, R., Ai, N., Xu,
D., and Fan, X. (2019). Copy number variation is
highly correlated with differential gene expression: a
pan-cancer study. BMC medical genetics, 20(1):175.
Shlien, A. and Malkin, D. (2009). Copy number variations
and cancer. Genome medicine, 1(6):1–9.
Simmons, S., Sahinalp, C., and Berger, B. B. (2016). En-
abling Privacy-Preserving GWASs in Heterogeneous
Human Populations. Cell Systems, 3(1):54 – 61.
Wang, S., Zhang, Y., Dai, W., Lauter, K., Kim, M., Tang,
Y., Xiong, H., and Jiang, X. (2016). HEALER: homo-
morphic computation of ExAct Logistic rEgRession
for secure rare disease variants analysis in GWAS.
Bioinformatics, 32(2):211–218.
Yoon, S., Xuan, Z., Makarov, V., Ye, K., and Sebat, J.
(2009). Sensitive and accurate detection of copy num-
ber variants using read depth of coverage. Genome
research, 19(9):1586–1592.
Zarrei, M., MacDonald, J. R., Merico, D., and Scherer,
S. W. (2015). A copy number variation map of the
human genome. Nature reviews genetics, 16(3):172–
183.
Zhang, F., Gu, W., Hurles, M. E., and Lupski, J. R. (2009).
Copy number variation in human health, disease, and
evolution. Annual review of genomics and human ge-
netics, 10:451–481.
Zhang, L., Bai, W., Yuan, N., and Du, Z. (2019). Com-
prehensively benchmarking applications for detecting
copy number variation. PLoS computational biology,
15(5):e1007069.
Privacy-preserving Copy Number Variation Analysis with Homomorphic Encryption
831
