
EnuwaJGX: Machine Learning Gene Prediction Software Application
Model - An Innovative Method to Precision Medicine and Predictive
Analysis of Visualising Mutated Genes Associated to Neurological
Phenotype of Diseases
Daniel F. O. Onah
a
Department of Information Studies, University College London, London, U.K.
Keywords:
Gene, Disease, Phenotype, Prediction, Mathematical Model, Machine Learning, Decision Tree, Search
Engine, Visualization.
Abstract:
This research investigates an aspect of precision medicine related to genes and their association with diseases.
Precision medicine is a growing area in medical science research. By definition precision medicine is an
approach that allows the selection of treatments that are most likely to help treat patients based on the genetic
understanding of their diseases. This approach proposes the customization of a medical model for healthcare,
treatment, medical decision making about genetic diseases and develop models that are tailored to individual
patient. There are readily available datasets provided by Genomics England related to diseases and the genes
that cause these diseases. This research presents a predictive technique that scores the possibilities of a mutated
gene causing a neurological phenotype. There are over a thousand genes associated with 26 subtypes of
neurological diseases as defined by Genomics England capturing genetic variation, gene structure and co-
expression network. The gene prediction was performed with search algorithms and methods that sequentially
looped through the database for true match. Linear search algorithm was applied along index search method to
perform the prediction matching of gene(s) that are associated to the disease(s). The prediction algorithm was
formulated based on a Mathematical/probabilistic concept that was used to design the model for processing
the data-set ready for gene prediction. It became apparent that over half a million (> 500, 000) genes were
predicted in this study that were associated to the neurological phenotype of the diseases in this research work.
1 INTRODUCTION
This study covers an innovative method to precision
medicine and predictive analysis of visualizing mu-
tated genes that are associated to neurological phe-
notype of diseases. A gene prediction study has re-
cently been explored in medical science research for
conducting gene analysis and how mutated genes are
associated to diseases.
The study applied a decision tree approach of ma-
chine learning and integrated this into the initial pre-
processing technique to generate each of the diseases
and their associated genes. This study’s hypothesis is
to predict that genes with higher frequency probabil-
ities of mutation in healthy individuals will be more
likely to cause the diseases. Previous studies revealed
that the ageing of the immune system of healthy adult
and the environment could be attributed to causation
a
https://orcid.org/0000-0001-6192-6702
of neurodegenerative diseases (Lagouge and Larsson,
2013; Sanchez-Guajardo et al., 2013).
This study covers primarily the area of Natural
Language Processing specifically in text segmenta-
tion and processing. A broader method was devel-
oped as a testable model to automatically analyse
datasets extracted from Genome England and other
medical data related to genes and diseases. The model
was used to explore the initial process of applying
NLP techniques and Machine learning approaches
such as text extraction, decision tree algorithm and
probability in precision medicine for conducting gene
search predictions. There are very few medical soft-
ware applications developed to perform this gene
search and prediction. This proposed research exten-
sion has led to the development of a gene application
system.
Gene prediction and expression has not been well
explored or done this way in medical research. This
Onah, D.
EnuwaJGX: Machine Learning Gene Prediction Software Application Model - An Innovative Method to Precision Medicine and Predictive Analysis of Visualising Mutated Genes Associated
to Neurological Phenotype of Diseases.
DOI: 10.5220/0011559100003335
In Proceedings of the 14th International Joint Conference on Knowledge Discovery, Knowledge Engineering and Knowledge Management (IC3K 2022) - Volume 1: KDIR, pages 281-291
ISBN: 978-989-758-614-9; ISSN: 2184-3228
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
281

proposed new way of presenting gene prediction de-
ficiencies or mutations using machine learning super-
vised and decision tree techniques that lead to specific
diseases is useful in making visualizing and search-
ing for these genes much easier. Applying Machine
Learning (ML) and a Natural Language Processing
(NLP) research approach is a novel ground in mod-
ern medical science research.
EnuwaJGX: Is a machine learning gene prediction
software web application model. It is a novel web
application model used for displaying search results
from a static data preprocessed for the gene predic-
tive analysis. The datasets stored in the web-database
captures innovative method to precision medicine and
also the visualisation of mutated genes that are associ-
ated to neurological phenotype of diseases. There are
several open source datasets that are related to neu-
rological phenotype of diseases and their associated
mutated genes that are available and provided by Ge-
nomics England
1
.
1.1 Research Hypothesis
• If a predicted gene has a probability greater than
or equal to the specified threshold, then the gene
is associated with the selected disease or diseases.
• If a predicted gene has a probability, less than the
specified threshold, then the gene is not associated
with the selected disease or diseases.
2 RELATED RESEARCH
Gene expression has been done with gene-specific
features capturing genetic variation, gene structure
and tissue-specific expression and co-expression. Pre-
viously, genes were randomly re-sampled with no
known disease association. Other studies have looked
at gene-phenotype predictions and generating genes
of distinct transcripts (Botía et al., 2018). One of the
main challenges of gene predictions is in finding an
optimal approach of combining the sources of infor-
mation extrinsic and intrinsic that could be associated
to gene mutation that are related to the genome of in-
terest (Bruna et al., 2020).
A study by Liu et al. (2018a) shows that thorough
approach in incorporating adequate evidence from
‘transcriptomes, machine learning tools, homology to
known sequences’ has proven challenging. Research
revealed that repetitive regions of gene sequences are
commonly a major issue when removed prior to gene
prediction. This sometimes led to multi-copy genes
1
https://www.genomicsengland.co.uk/
being misidentified as repeats in the analysis and ex-
cluded from the final result predictions (Chen et al.,
2020, 2019).
A related research study identified one of the
causative factors of gene mutation is as the result of
alteration of the ‘immunosenescence’ defence mecha-
nism caused be ageing (Chong et al., 2003).
2.1 Data Representativeness
According to Sikibi (2022), the first step of data
preparations is the collection, exploration and discov-
ery of all the data from the potential sources and iden-
tify patterns, relationships, annotation, missing data
features, etc. The data cleaning process helps to iden-
tify errors within the datasets which could be cor-
rected in the process of restructuring and formatting
the datasets. According to de Araujo et al. (2022),
data cleaning includes removing duplicated data, un-
wanted data, making unstructured data to structured
format and filtering-out any personal data that would
not be relevant for the analysis. This study applied
a systematic data preprocessing and representative-
ness approach. Mathematical and probability algo-
rithms were constructed using Python programming
language and algorithm models to perform this data
representativeness and preprocessing.
3 RESEARCH METHODS
In this section, the research model is described. The
model in this study is to illustrate the hypothesis and
support the implementation of the system to answer
the study’s research questions.
Gene prediction is one of the most fascinating
problem to solve in computational biology (Flicek,
2007). In the study, supervised Machine leaning tech-
nique was applied to generate disease-gene specific
classifiers. Decision tree classifiers were applied at
the initial data-set collection process. A supervised
machine learning(ML) approach was used to create a
model of gene phenotype revealing the associations of
the generated classifiers that could be classified and
distinguished between genes that have relative asso-
ciation with other neurological phenotype and those
that do not. Genes that show this relationship to spe-
cific neurological phenotype are generally linked to
the causation of the disease than those with no di-
rect association to the neurological phenotype (Botía
et al., 2018).
KDIR 2022 - 14th International Conference on Knowledge Discovery and Information Retrieval
282

3.1 Supervised Learning
Several machine learning applications require a
model in order to make accurate predictions on test
data samples that are distributionally different from
training dataset. In order to enable web based elec-
tronic data prediction, there is a need for automated
methods of data analysis. Machine learning provides
these developing methods that can automatically de-
tect patterns in data and then use the uncovered pat-
terns to perform data prediction (Murphy, 2012). This
research applied a probabilistic approach of super-
vised learning to data processing, analyses and pre-
diction. Several supervised machine learning meth-
ods have been widely used to predict disease genes
(Asif et al., 2018). Gene expression data required
high-capacity of machine learning methods that are
capable of identifying disease association (McDer-
mott et al., 2019) and developing models to perform
related gene prediction to identify disease candidate
bio-markers in gene classification (Lyu and Haque,
2018).
3.2 Mathematical Model & Probability
Algorithm
In order to formulate the domain, a special mathemat-
ical model was developed for the data processing that
considered the limitations of the datasets and original
structures. The model introduces the basic terms, spe-
cific features from the processed data. This study pro-
poses a probabilistic model for data pre-processing,
normalisation and transformation (Lai et al., 2004)
that enabled the identification of genes that are closely
associated to diseases (see equation 1).
Prob
pred
=
n
∑
i
count(T P
cl
)
count(T P
cl
) + count(FP
cl
)
(1)
where
• TP = True positive
• FP = False positive
• cl = Decision tree classifier
3.3 Hypothesis Testing
Hypothesis. In order to decide the precision of the
gene prediction, we applied the if single-selection
statement approach within the algorithm.This ap-
proach allows us to choose among the alternative
course of actions that would successfully meet the
condition of our gene and disease predictive associ-
ation. In this study, the threshold decided was 0.99
(99% confident interval). The equation applied within
the algorithm predicts the gene(s) that are associated
to the disease(s) if the threshold condition is true and
otherwise non-association to the disease(s) (as seen in
2 and 3).
Disease
pred
= Prob
pred
≥ Prob
T hreshold
(2)
Non − Disease
pred
= Prob
pred
< Prob
T hreshold
(3)
The threshold mark when varies may affect the
precision accuracy of the gene prediction. In order to
reach a more accurate prediction, the threshold mark
decided should be closer to a perfect probability. In
this case, we considered 0.99 to 1 (99% - 100%) as
the precision probability for a perfect gene prediction
in this study.
Precision. In this study, precision within the gene
prediction is very important for the accuracy of the
final predictions. Precision was considered based on
the number of true positive over or divided by the total
of true positive and false positive (equation 4).
Precision =
∑
(
T P
T P + FP
)
(4)
TP: This is the true positive Boolean value in the
dataset that represent the frequency count of the true
variable response from the decision tree classifiers.
FP: This is the false positive Boolean value in the
dataset that does not include the true response but
false within the decision tree classifiers of the dataset.
In processing the data, the first two columns of the
dataset were excluded from the pre-processing stage
as these contains the serial numbers and the gene
columns. The equation 5 below shows how this was
avoided.
Precise
rn
=
∑
(Rn) − 2 (5)
Rn is the number of classifier in a data row in the
dataset.
The above equation was introduced to calculate
the precise classifier in the dataset row by avoiding
the first two columns. This is then fed into the next
equation 6 for the accurate pre-processing of the gene
prediction within the dataset.
Precision
prob
=
∑
T P
Precise
rn
(6)
This equation calculates and processes the data
of interest without the first two columns and rows
within the dataset. We are only interested in the 200
EnuwaJGX: Machine Learning Gene Prediction Software Application Model - An Innovative Method to Precision Medicine and Predictive
Analysis of Visualising Mutated Genes Associated to Neurological Phenotype of Diseases
283

responses of the decision tree classifier. We do not
need to process these first two columns because they
contain the serial number in the first column and the
genes in the second column.
3.4 Gene Annotation
Each gene is annotated separately within the associ-
ated disease files. This individual gene annotation
makes the search for mutated genes and its associa-
tion to be possible within the disease database. This
process enable the serialization of the gene prediction
data and the precision accuracy of the output predic-
tion. For example, in Table 1 , UQCRC1 is identified
as a mutated gene likely to cause neurological pheno-
type of Allinone disease. Suppose, there are no gene
annotation, then the precision of the gene result pre-
diction could have been hampered in this study. The
annotation of mutated gene association led to proba-
bilistic prediction of genes and association or causa-
tion leading to a disease. This process of gene annota-
tion is performed in sequential order during each data
processing stages. During the gene search, the pro-
gram retrieve predicted gene data sequentially from
the data-set by starting from the beginning of the file
and reading all the data consecutively until the desired
gene(s) information and association is found. The ac-
tion applied is to identify the true gene value in the
data-set that are associated to the disease(s) and not
taking into consideration the false value.
3.5 Linear Search Method
This research applied linear search methods for the
prediction of the processed data-set. The linear search
algorithm searches each gene in the database sequen-
tially. If the search key is associated to a gene then
there will be a true prediction. However, if the search
key does not match any gene after reaching the end of
the data-set stored in the database then this will output
a statement that the search gene is not found. There-
fore, the search algorithm determine that the gene key
entered does not match any gene sequentially and in
this case returns no result. In addition, if the search
gene is found in the database, the algorithm tests and
compares all genes associated with the search gene
until it finds one or all that matches the search and
returns that gene(s) and the associated disease(s).
3.6 Index Search Method
Another search method that was used in the early
stage of the search engine development was the in-
dex search method. This method was considered as
well because of it’s similarity with the linear search
method. The algorithm receives a gene to search for
from the database and the search key. The algorithm
loops through the genes in the database and compares
each with the search gene key. If the values are equal
or true is return, then the algorithm returns the index
of the search gene. However, if there are duplicate
genes in the database, the search returns the index of
the first gene in the database sequentially that matches
the search key. But, if the search ends without any
true match return, then the algorithm will output no
gene found or processed in the database.
3.7 Raw Dataset
The dataset for this research are in an unstructured
state that need to be processed. These are made up
of 26 Neurological Phenotype of diseases. These are
made up of combination of Boolean values of True
and False distributed in the rows and columns of the
datasets. The rows are made up of over >17,000
genes and columns hold 200 decision tree classifiers
for each disease file as shown in the sample Figure 1.
Figure 1: Raw dataset with decision tree Boolean classi-
fiers.
3.8 Prediction
In order to make an accurate prediction in this study,
a threshold of 0.99 (99%) was decided so as to closely
relate to a perfect probability to perform the final gene
prediction. Here, we use the calculation of the pre-
cision probability to perform the prediction as illus-
trated in the above (equation 4).
Similarity Prediction. In this study, similarity was
performed between disease predictions. The result
from gene predictions associated to specific selected
diseases was used as the control sample to perform
search prediction of the other diseases. The equation
7 below allows the selection of the first 10 genes that
passed the threshold in ascending other. Within these
10 genes, selection of those that met perfect probabil-
ity of the value 1 (one) are used to search the database
for whether they are associated to the other remaining
KDIR 2022 - 14th International Conference on Knowledge Discovery and Information Retrieval
284

diseases in the database. Within the study, selection of
two diseases were used as the control or gold standard
experiment to conduct the similarity search as seen in
Table 1 and Table 2.
Precision
prob
≥ T hreshold
(7)
Where:
Threshold = 0.99
The mathematical model and algorithm was trans-
lated into a simple Python programming construct as
illustrated in screenshot image in Figure 2 for the pre-
processing and prediction of the results.
Figure 2: Mathematical model and algorithm transformed
into python construct for preprocessing the data and gene
prediction.
4 RESEARCH PIPELINE
Figure 3 illustrates the various sequential steps intro-
duced in the data processing pipeline for this study. In
order to process the data for accurate supervised gene
predictions, the steps within the pipeline must be se-
quentially followed, that is from one preceding step
to the other.
Figure 3: Machine Learning data processing pipeline.
Data Collection: The datasets for this study are made
up of 26 unstructured neurological phenotype dis-
ease data with 200 decision tree classifiers that were
used to classify the response information contained in
it. These data were extracted from Genome England
database. Some of the datasets are currently available
as open source.
Figure 4 is the flowchart that illustrates the stages
involved in the data processing and prediction within
this research study.
Figure 4: Flowchart algorithm illustrating the data process-
ing method and predictions.
Preparation and Preprocessing: Although, the
dataset that was extracted were in an unstructured
state, a probabilistic algorithm was designed to per-
form the initial preprocessing and cleaning of the data
for easy extraction of relevant genes for the predic-
tions.
Figure 5 presents the predicted result from the pre-
processed raw dataset. There are over half-a-million
(> 500,000) genes predicted in this study. These re-
sults are stored in the PostgrSQL database for the Ma-
chine Learning gene prediction web-based applica-
tion designed using Python−D jango application pro-
gramming interface (API) framework model as pre-
sented in this study.
Feature Extraction: The next process was to per-
form feature extraction as related to genes and dis-
eases. Feature extraction was the second to the last
processes before the gene predictive analysis and data
visualisation was done. In this step, genes of interest
were selected from the pre-processed data that met the
condition of the stringency threshold specified in the
study.
Prediction: This allowed the extraction of common
genes in association with multiple diseases that could
help in the visualisation of the results.
Visualisation: This was the final process of the
pipeline, which then led to the research results, analy-
sis and visualisation of the predictions. The predicted
results can be downloaded as comma-separated val-
ues(CSV) and also as images.
EnuwaJGX: Machine Learning Gene Prediction Software Application Model - An Innovative Method to Precision Medicine and Predictive
Analysis of Visualising Mutated Genes Associated to Neurological Phenotype of Diseases
285

Figure 5: A sample of a preprocessed dataset gene predic-
tion.
4.1 Control Disease for Experiment
Two neurological disease samples were selected from
26 diseases for this experiment as indicated in Table
1 and Table 2.
Table 1: Allinone disease as gold-standard control experi-
ment I.
Allinone disease
Gene Prediction
UQCRC1 1.000
KCNMA1 1.000
ABCC9 1.000
ACACB 1.000
PDLIM5 1.000
CEP192 1.000
WWC2 1.000
IMMT 1.000
BZW2 0.995
DST 0.995
Table 2: Parkinson disease as gold-standard Control exper-
iment II.
Parkinson disease and complex Parkinsonism
Gene Prediction
SLC41A2 1.000
IMMT 0.995
AKAP6 0.995
MED24 0.990
CD22 0.990
VPS13C 0.990
MEIS2 0.990
RAP1GDS1 0.990
PAM 0.990
ALAD 0.990
5 RESULTS & FINDINGS
Table 3 describes the predicted data from the machine
learning gene prediction model. These predictions are
based on the probability of classifiers that was pre-
dicted. The gene search for is either associated to a
disease(s) or not. The table shows and describes fur-
ther statistical analysis to present the standard devia-
tion, the minimum and maximum gene prediction and
the interquartile range of 25%, 50% and 75%.
Table 3: Mutated Genes standard deviation.
Probability of classifiers that voted "disease" or "non-disease"
count 121.000000
mean 0.525702
std 0.255633
min 0.040000
25% 0.360000
50% 0.540000
75% 0.720000
max 0.990000
Figure 6 describes further the result by illustrat-
ing the probability level of the mutated genes using a
histogram.
Figure 6: Described the frequency of the search genes.
KDIR 2022 - 14th International Conference on Knowledge Discovery and Information Retrieval
286

5.1 Mutated Genes and Associated
Diseases
This section illustrates the visualization of mutated
genes that are associated with the neurological phe-
notype of diseases in this study. Preliminary stud-
ies revealed that using the control genes from experi-
ment I and II on the machine learning web-based ap-
plication model helped to understand whether there
are any correlations or similar results from other dis-
eases within the database model. In the first con-
trol experiment, the study applied the full thresh-
old of 0.99 probability. The result is displayed in
Figure 7. The 10 most genes associated with Alli-
none disease, include UQCRC1, KCNMA1, ABCC9,
ACACB, PDLIM5, CEP192, WWC2, IMMT, BZW2,
DST. However, gene IMMT is also associated with
Parkinson and Complex Parkinsonism disease but
with different level of predictions. This predictive
level comparison indicated that gene IMMT is more
directly associated to Allinone disease as compare to
Parkinson and complex Parkinsonism disease as seen
in the Table 1 and Table 2 control experiment results.
Figure 7: Results showing other diseases with similar mu-
tated genes that were associated to allinone disease.
In the second control experiment II, we applied the
same threshold stringency probability of 0.99 within
the database model in order to observe the result.
In this study, our chosen assumption of good mea-
sure probability was from 0.99 and above. Both con-
trol experiments revealed some similarities between
genes mutation within other neurological phenotype
diseases within this study. The study was able to
predict genes that are related to each other within
several diseases. Figure 8 shows the results from
the control experiment II. The 10 most genes asso-
ciated with Parkinson and Complex Parkinsonism
disease, include SLC41A2, IMMT, AKAP6, MED24,
CD22, VPS13C, MEIS2, RAP1GDS1, PAM, ALAD
as seen in Table 2. Parkinson disease causation is
widely associated to ageing immune cells (Lagouge
and Larsson, 2013) and the environmental factors that
could lead to the gene mutation in healthy humans
(Niccoli and Partridge, 2012). There are also several
pathogenic missense mutations that segregate in fam-
ilies that could also be associated to Parkinson’s dis-
ease (Paisán-Ruız et al., 2004; Zimprich et al., 2004;
Liu et al., 2018b).
Figure 8: Results showing other diseases with similar mu-
tated genes that were associated to Parkinson and Complex
Parkinsonism disease.
Table 4 shows a few selected diseases with similar
mutated genes from the results of the control experi-
ment I and II in this study.
6 HARD COMPUTATION
PROBLEM
Although several tests were performed on the algo-
rithm model to ascertain the performance and the pre-
cision accuracy of the gene prediction model. One
of the most difficult challenges of this machine learn-
ing model is designing an optimization search engine
mechanism that will enable the precision of the gene
prediction search from the database.
6.1 Machine Learning Web-App: Gene
Search Engine Optimization
In the study, a proposed machine learning web-app
known as EnuwaJGX was developed to allow the
searching of predicted genes that are associated to
these diseases. The users are able to enter multiple
genes as input into the gene application web search in-
terface, separated with a comma and all capital letter
entry. The flowchart in Figure 9 illustrates the process
of searching and the display of only relevant genes
that are associated with these diseases. In this study,
we are only interested in the genes that are mutated
and that causes these neurological phenotype of the
diseases. Further research work will be conducted us-
ing natural language processing (NLP) techniques to
explore more of the predicted genes that are associ-
ated with multiple diseases in the future study.
EnuwaJGX: Machine Learning Gene Prediction Software Application Model - An Innovative Method to Precision Medicine and Predictive
Analysis of Visualising Mutated Genes Associated to Neurological Phenotype of Diseases
287

Table 4: Mutated Genes from control experiment I & II revealing similarities with a few selected diseases.
Disease Gene (from control Exp. I) Disease Gene ( from control
Exp. II)
Epileptic En-
cephalopathy (EE)
KCNMA1 Early Onset Dementia Encompass-
ing Fronto Temporal Dementia and
Prion Disease (EODM FTD PrD)
MEIS2, ALAD
Congenital Myopa-
thy (CMP)
UQCRC1, ACACB, IMMT,
PDLIM5, ABCC9, KCNMA1
Epileptic Encephalopathy (EE) MED24, CD22,
MEIS2, PAM
Skeletal Muscle
Channelopathies
(SMC)
ACACB Congenital Myopathy (CMP) VPS13C, IMMT,
MEIS2, PAM,
AKAP6
Inherited White
Matter Disorders
(IWMD)
BZW2 Congenital Muscular Dystrophy
(CMD)
MED24
Epilepsy Plus (EP) KCNMA1 Hereditary Ataxia (HA) MED24
Limb Girdle Mus-
cular Dystrophy
(LGMD)
PDLIM5 Inherited White Matter Disorders
(IWMD)
IMMT, AKAP6
Arthrogryposis
(AMC)
ACACB, ABCC9 Epilepsy Plus (EP) MED24, CD22,
MEIS2, CD22,
MEIS2, PAM
Rhabdomyolysis and
Metabolic Muscle
Disorders(RMMD)
ACACB, IMMT, PDLIM5, ABCC9 Limb Girdle Muscular Dystrophy
(LGMD)
MED24, IMMT,
AKAP6
Structural Basal
Ganglia Disorders
(SBGD)
PDLIM5 Allinone (A) VPS13C, IMMT,
MEIS2, PAM,
AKAP6
Congenital Myaes-
thenia (CMS)
ACACB Early Onset Dystonia (EODS) MED24,
CD22, IMMT,
MEIS2,SLC41A2,
RAP1GDS1,PAM,
ALAD
Distal Myopathies
(DM)
UQCRC1, ACACB, IMMT,
BZW2, PDLIM5, ABCC9, KC-
NMA1
Hereditary Spastic Paraplegia
(HSP)
MED24
- - Arthrogryposis (AMC) VPS13C, IMMT
- - Rhabdomyolysis and Metabolic
Muscle Disorders(RMMD)
MED24, CD22,
VPS13C,
IMMT,RAP1GDS1,
PAM, AKAP6
- - Brain Channelopathy (BC) RAP1GDS1,
AKAP6
- - Structural Basal Gangila Disorders
(SBGD)
MED24, IMMT,
MEIS2, SLC41A2,
AKAP6
- - Cerebrovascular Disorders (CD) MEIS2,RAP1GDS1,
PAM
- - Intracerebral Calcification Disor-
ders (ICD)
SLC41A2
- - Distal Myopathies (DM) MED24,
IMMT, MEIS2,
RAP1GDS1,PAM,
AKAP6
- - Charcot Marie Tooth Disease VPS13C, MEIS2
KDIR 2022 - 14th International Conference on Knowledge Discovery and Information Retrieval
288

Initialize
Database
Model
Search
Input Gene
Disease
End
Figure 9: Flowchart illustrating the gene search process.
Figure 10 illustrate the overarching search op-
timization process and the return mechanism that
present a notification that a gene search for does not
exist in the database or has not been in the list of pro-
cess genes for the study. This provide the user with
another opportunity to perform a new gene search ac-
tion and the system will then optimise the search en-
gine from the beginning following through the same
linear search process until a true search result is iden-
tified or the search process ends.
Figure 10: Client application interface search process.
6.2 Web App: EnuwaJGX
EnuwaJGX
2
is a web-based tool that allows life
scientists, biomedical researchers, system biologist,
bioinformaticians, data scientists and medical sci-
ences professionals to take advantage of this novel
machine learning gene prediction application with
powerful function and class of techniques in an appro-
priate user interface. Large complex models of data
processing and storage are built and analysed through
a graphical user interface. It is an ongoing tool devel-
opment for research to allow the large scale prediction
of genes and their associated diseases. Results from
the search genes and associated diseases can be down-
2
http://35.176.149.24:8000/gene/
loaded freely from the application database to enable
researchers conduct further analysis. The future plans
is to allow for the analysis of genome-scale gene and
disease models visualization.
The Machine Learning System Design Architec-
ture: Figure 11 describes the flow diagram and con-
struct of the Machine Learning web-based application
architecture for this study.
Figure 11: Machine Learning system application design ar-
chitecture.
6.3 EnuwaJGX System Application:
Testing & Evaluation
The EnuwaJGX web-based software application sys-
tem has been tested and evaluated for the validity of
the predictions by Clinicians, Geneticist, Biologist
and Bioinformaticians. They were all able to iden-
tify gene causation and predictions associated to the
neurological phenotype of diseases for which they are
investigating. A cross testing of expert review on mu-
tated genes and diseases causation was conducted us-
ing the datasets from PanelApp (325 panels)
3
. This
evaluation reviewed similar predictive genes that are
associated to the subtypes of neurological phenotype
of diseases predicted in the EnuwaJGX application
system. We conducted validity of our gene prediction
application results with other existing expert verified
results. For example, testing the gene ACT B which
has been reviewed to be the top cause of Early onset
dystonia disease in the PanelApp. The EnuwaJGX
system also predicted the same result with 95% con-
fident interval, revealing that the mutation of ACT B
gene caused the Early onset dystonia disease.
7 CONCLUSION
This study used a Machine Learning model to pre-
dict genes that are directly associated to known dis-
eases. The findings presented results from the gene
3
https://panelapp.genomicsengland.co.uk/panels/
EnuwaJGX: Machine Learning Gene Prediction Software Application Model - An Innovative Method to Precision Medicine and Predictive
Analysis of Visualising Mutated Genes Associated to Neurological Phenotype of Diseases
289
predictions that revealed disease causation genes. The
datasets were able to show relative genes that are as-
sociated to specific neurological diseases.
The predictions from this research has shown dis-
tinctive gene predictions and predict distinct genes
closely associated to the selected diseases.
The most interesting and novel area for further
study is to apply Natural Language Processing tech-
niques to develop a different model with new sources
of data and compare the findings with the previous re-
sults of the ground truth from the research of the Ma-
chine Learning(ML) and probabilistic models used
to predict the genes associated to diseases in this
study. By combining these two research approaches
and conducting the comparisons, this will provide us
with a more clear understanding of the accuracy of the
gene predictions.
This research has associated mutated genes causa-
tion to neurodegenerative phenotype of diseases. One
of the main factors of gene mutation was associated
to both ageing, environment and life style of humans.
Further research will be done on applying NLP
and ML approach on the most recent medical datasets
(text based) related to genes and diseases.
7.1 PubMed-PMC Key Term Search
Initial searching of PubMed central - PMC (US Na-
tional Library of Medicine National Institutes of
Health) databases using key words of genes and dis-
eases produces over 600 and 20,000 journal papers
respectively. This proposed extended research will
compare the analysis of the ground truth as defined
from within the extracted articles and the initial gene
prediction results from the machine learning gene pre-
diction model in order to confirm the correlation and
similarities within the datasets. This extended re-
search will be applying a TextRank algorithm, which
is popular for conducting similarity matrix of mul-
tiple datasets extraction and automated summariza-
tion. In this proposed phase of the research, the study
will be using this algorithm within the extracted jour-
nal datasets from PubMed-PMC and the previous Ge-
nomic datasets analysed from the Phase 1 of this re-
search.
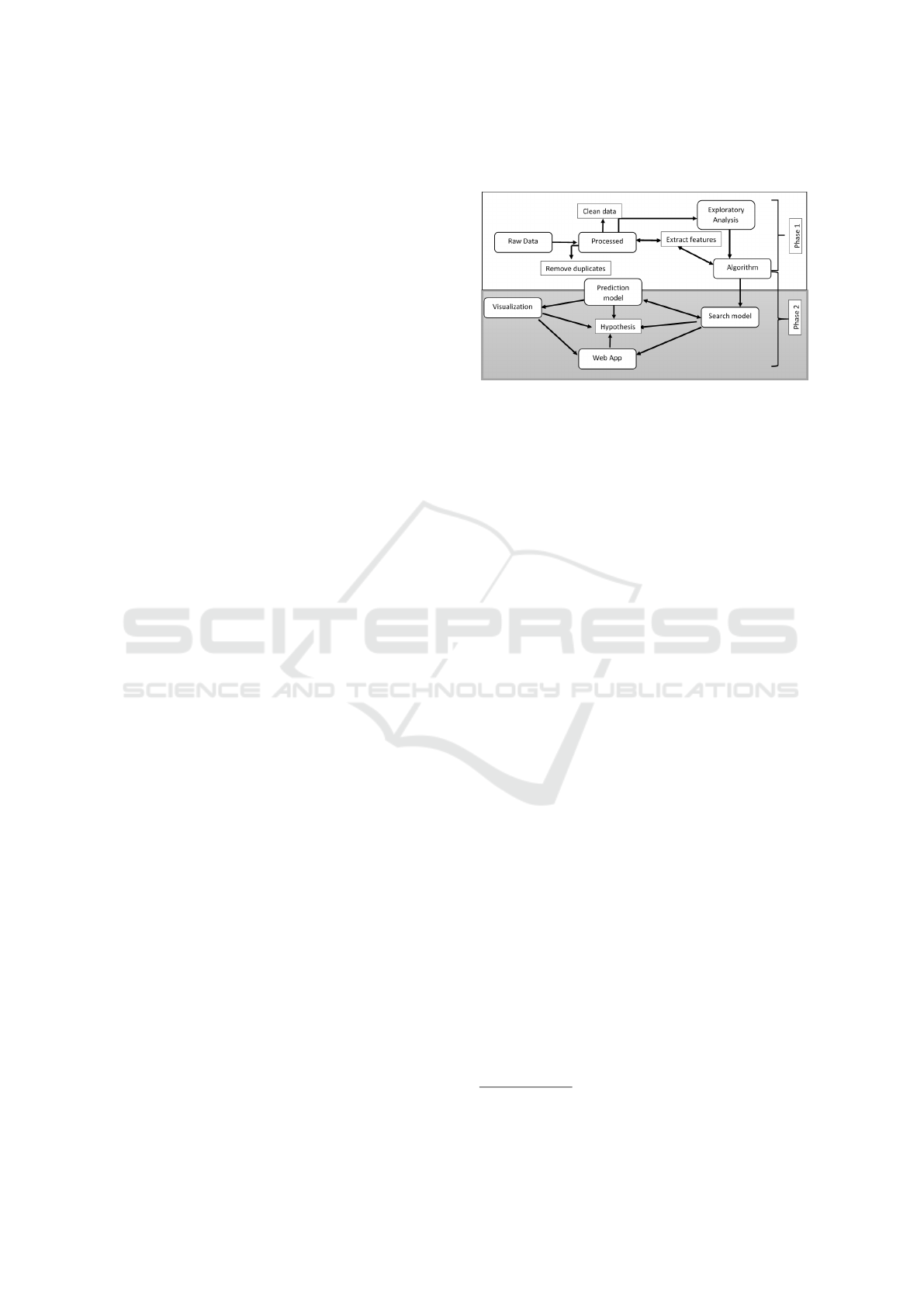
7.2 Extended Research Pipeline
The early phase of this extended research pipeline ar-
chitecture has been completed from a computer pro-
gramming ‘code perspective’, that enabled the initial
gene predictions using the console of the operating
system. The back-end and interface is the next ongo-
ing proposed Phase 2. This Phase will continuously
apply the proposed hypothesis in the system design
(see Figure 12).
Figure 12: Full extended gene prediction pipeline.
REPRODUCIBILITY AND ONLINE
RESOURCES
Data and Code Availability
The datasets analysed in this study with all the codes
for the data preprocessing, model implementation, de-
sign web-based deployment architecture for the the
disease and gene prediction and all algorithms are
made available at a GitHub repository
4
. Additionally,
this repository also contains the full web development
using Django platform that supported the Python pro-
gramming code and the database that was restructured
or converted from SQLite3 to PostgreSQL in order
to store the large volume of the processed gene and
disease data for the study. The author is fully com-
mitted to maintaining continuously the repository in
terms of both updating with new models or codes that
changes the performance, functionality and ease of
use of the EnuwaJGX website, and actively monitor-
ing any issues within GitHub. EnuwaJGX Machine
Learning genes and diseases prediction web resources
and database are available within the GitHub reposi-
tory.
AUTHORS’ CONTRIBUTIONS
This research was conducted by a single author. The
author analysed and processed the datasets retrieved
from Genomics England database. All Mathemati-
cal algorithm models and code for the preprocessing
of the datasets were developed solely by the author.
The author developed a web-based semantic software
4
https://github.com/DanFON/machinelearningpredict
gene/
KDIR 2022 - 14th International Conference on Knowledge Discovery and Information Retrieval
290

application system for the gene and disease predic-
tions. The software application model was devel-
oped using Python − D jango platform environment
and a PostgreSQL database. This can be found in the
EnuwaJGX web-based application. The findings and
results from this research were analysed by the main
author of this paper.
ACKNOWLEDGEMENTS
The Author wish to acknowledge funding from the
JWECT for awarding the grant to continue this re-
search. Thanks to UCL Institute of Neurology for
testing the prototype Machine Learning gene predic-
tion web-based application and Genomics England
for providing the initial Dataset. A big thank you to
Dr Elaine Pang for proofreading the manuscript.
REFERENCES
Asif, M., Martiniano, H. F., Vicente, A. M., and Couto,
F. M. (2018). Identifying disease genes using machine
learning and gene functional similarities, assessed
through gene ontology. PloS one, 13(12):e0208626.
Botía, J. A., Guelfi, S., Zhang, D., D’Sa, K., Reynolds, R.,
Onah, D., McDonagh, E. M., Martin, A. R., Tucci,
A., Rendon, A., et al. (2018). G2p: Using machine
learning to understand and predict genes causing rare
neurological disorders. bioRxiv, page 288845.
Bruna, T., Lomsadze, A., and Borodovsky, M. (2020).
Genemark-ep+: eukaryotic gene prediction with self-
training in the space of genes and proteins. NAR Ge-
nomics and Bioinformatics, 2(2):lqaa026.
Chen, Y., González-Pech, R. A., Stephens, T. G., Bhat-
tacharya, D., and Chan, C. X. (2020). Evidence
that inconsistent gene prediction can mislead analy-
sis of dinoflagellate genomes. Journal of phycology,
56(1):6–10.
Chen, Y., Stephens, T. G., Bhattacharya, D., González-
Pech, R. A., and Chan, C. X. (2019). Evidence that
inconsistent gene prediction can mislead analysis of
algal genomes. bioRxiv, page 690040.
Chong, Y., Ikematsu, H., Yamaji, K., Nishimura, M., Kashi-
wagi, S., and Hayashi, J. (2003). Age-related accumu-
lation of ig vh gene somatic mutations in peripheral b
cells from aged humans. Clinical & Experimental Im-
munology, 133(1):59–66.
de Araujo, A. L., Hardell, C., Koszek, W. A., Wu, J., and
Willemink, M. J. (2022). Data preparation for artifi-
cial intelligence. In Artificial Intelligence in Cardio-
thoracic Imaging, pages 37–43. Springer.
Flicek, P. (2007). Gene prediction: compare and contrast.
Genome biology, 8(12):233.
Lagouge, M. and Larsson, N.-G. (2013). The role of mi-
tochondrial dna mutations and free radicals in disease
and ageing. Journal of internal medicine, 273(6):529–
543.
Lai, Y., Wu, B., Chen, L., and Zhao, H. (2004). A sta-
tistical method for identifying differential gene–gene
co-expression patterns. Bioinformatics, 20(17):3146–
3155.
Liu, H., Stephens, T. G., González-Pech, R. A., Beltran,
V. H., Lapeyre, B., Bongaerts, P., Cooke, I., Aranda,
M., Bourne, D. G., Forêt, S., et al. (2018a). Sym-
biodinium genomes reveal adaptive evolution of func-
tions related to coral-dinoflagellate symbiosis. Com-
munications biology, 1(1):1–11.
Liu, Z., Bryant, N., Kumaran, R., Beilina, A., Abeliovich,
A., Cookson, M. R., and West, A. B. (2018b). Lrrk2
phosphorylates membrane-bound rabs and is activated
by gtp-bound rab7l1 to promote recruitment to the
trans-golgi network. Human Molecular Genetics,
27(2):385–395.
Lyu, B. and Haque, A. (2018). Deep learning based tumor
type classification using gene expression data. In Pro-
ceedings of the 2018 ACM international conference
on bioinformatics, computational biology, and health
informatics, pages 89–96.
McDermott, M. B., Wang, J., Zhao, W.-N., Sheridan,
S. D., Szolovits, P., Kohane, I., Haggarty, S. J., and
Perlis, R. H. (2019). Deep learning benchmarks on
l1000 gene expression data. IEEE/ACM transac-
tions on computational biology and bioinformatics,
17(6):1846–1857.
Murphy, K. P. (2012). Machine learning: a probabilistic
perspective. MIT press.
Niccoli, T. and Partridge, L. (2012). Ageing as a risk factor
for disease. Current biology, 22(17):R741–R752.
Paisán-Ruız, C., Jain, S., Evans, E. W., Gilks, W. P., Simón,
J., Van Der Brug, M., De Munain, A. L., Aparicio,
S., Gil, A. M., Khan, N., et al. (2004). Cloning of
the gene containing mutations that cause park8-linked
parkinson’s disease. Neuron, 44(4):595–600.
Sanchez-Guajardo, V., Barnum, C. J., Tansey, M. G., and
Romero-Ramos, M. (2013). Neuroimmunological
processes in parkinson’s disease and their relation to
α-synuclein: microglia as the referee between neu-
ronal processes and peripheral immunity. ASN neuro,
5(2):AN20120066.
Sikibi, M. (2022). Use data mining cleansing to prepare
data for strategic decisions. In Data Mining-Concepts
and Applications. IntechOpen.
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer,
M., Lincoln, S., Kachergus, J., Hulihan, M., Uitti,
R. J., Calne, D. B., et al. (2004). Mutations in lrrk2
cause autosomal-dominant parkinsonism with pleo-
morphic pathology. Neuron, 44(4):601–607.
EnuwaJGX: Machine Learning Gene Prediction Software Application Model - An Innovative Method to Precision Medicine and Predictive
Analysis of Visualising Mutated Genes Associated to Neurological Phenotype of Diseases
291
